UNIVERSITA' DEGLI STUDI DI MILANO
Facoltà di Scienze Matematiche, Fisiche e Naturali
Corso di Laurea in Scienze Biologiche

MODIFICAZIONI DELLA REGOLAZIONE GENICA INDOTTE DALLO STRESS CRONICO IN UN MODELLO PRECLINICO DI DEPRESSIONE: EFFETTI DELL'ANTIPSICOTICO ATIPICO QUETIAPINA
Relatore: Dott.ssa Mariaelvina SALA
Correlatore: Prof. Marco ORSETTI
Tesi di laurea di:
Federica PODETTA
Matr. n. 512572
Anno Accademico 2005-2006
"Non c'è una cellula, un atomo,
una virgola che possa sfuggire
all'unità del cosmo.
C'è ovunque il segno di Dio.
.il segno non cesserà di
Interrogarmi."
a mamma e papà
a Massimo
INDICE
INTRODUZIONE
I disturbi affettivi
Il disturbo depressivo maggiore
1.1.2 Il disturbo bipolare
Neurobiologia della depressione
Ipotesi monoaminergica
Oltre l'ipotesi monoaminergica
1.2.3 Ruolo dello stress nei disturbi affettivi
Test preclinici per la valutazione dell'attivita' degli antidepressivi
Asportazione dei bulbi olfattivi
Test del nuoto forzato o test di porsolt
Learned helplessness
Stress cronico variato
1.3.5 Ratti con fenotipo swlo e swhi
Gli antipsicotici atipici nel trattamento dei disturbi affettivi
Definizione di antipsicotico atipico
1.4.2 Basi farmacologiche dell'atipicità
1.4.3 Clozapina
1.4.4 Olanzapina
Risperidone
1.4.6 Quetiapina
SCOPO DELLA TESI
MATERIALI E METODI
Stress cronico variato (cms)
Stabulazione e manipolazione degli animali
Scelta dei ratti preferenti al saccarosio
Induzione dell'anedonia
Valutazione dello stato anedonico
Trattamento e prelievo delle aree
3.1.6 Estrazione di rna
DNA microarray
RT-PCR real-time
3.3.1 Preparazione primers per il settaggio della real-time
3.3.2 Preparazione della curva di taratura
3.3.3 Preparazione dei campioni
RT-PCR semiquantitativa
3.4.1 Retrotrascrizione
Amplificazione
Corsa elettroforetica
RISULTATI
DISCUSSIONE
BIBLIOGRAFIA
Il Manuale Diagnostico e Statistico delle Malattie Mentali (DSM-IV TR) descrive due disturbi dell'umore, il disordine depressivo maggiore o disturbo unipolare e il disturbo bipolare, suddivisi a loro volta in diversi sottotipi (Kandel e coll. 1994).
Il disordine depressivo maggiore (DDM) è uno dei disturbi psichiatrici più diffusi e la sua frequenza negli ultimi decenni sembra essere in costante aumento, non solo nella popolazione adulta e anziana, ma anche tra gli adolescenti.
Il picco di comparsa della malattia è compreso tra i 20 e i 40 anni di età. Il DDM è una patologia disabilitante che colpisce il 7-15% degli uomini e il 13-28% delle donne, esercitando importanti effetti sul comportamento individuale e sociale e soprattutto sulla capacità del soggetto malato di operare normalmente all'interno della società.
Tale disturbo rappresenta una fonte significativa di sofferenza per i pazienti che ne sono afflitti e per i loro familiari, in quanto tende a cronicizzare e può durare per l'intero arco della vita (Weisman e coll. 1998).
Come definito dall'APA, l'American Psychiatric Association, il DDM è una patologia eterogenea, che si manifesta spesso con sintomi che abbracciano l'area psicologica, comportamentale e fisiologica. Pertanto la diagnosi è affidata alla valutazione di un'ampia gamma di manifestazioni cliniche. La diversità dei sintomi, variabili per definizione, interessa l'ambito emotivo-affettivo (ricorrenti sentimenti di colpa e autoriduttivi, diminuzione dell'autostima), le funzioni vegetative (disturbi del ciclo sonno-veglia, dell'appetito e del comportamento sessuale), l'ambito psicomotorio (eloquio alterato, agitazione psicomotoria e iperattività o rallentamento e a 515g621f patia) e la sfera cognitiva (disturbi della memoria, minore capacità di concentrazione ed attenzione, ricorrenti pensieri di morte e ideazione suicidaria). E' stato stimato che il suicidio costituisca la causa di morte del 15% dei pazienti depressi.
Il DDM rappresenta perciò una sindrome multifattoriale ed eterogenea (Rossi, Cuomo e Riccardi, 2005) che può provocare patologie di interesse medico generale. Alcuni autori sostengono infatti che il DDM rappresenti uno dei maggiori rischi di insorgenza di patologie cardiovascolari (Starkstein e coll. 1989). Inoltre sintomi depressivi, difficilmente distinguibili dalla depressione maggiore, si verificano in svariate condizioni mediche come disturbi endocrini, disturbi vascolari, morbo di Parkinson, alcuni tumori, diabete e ictus (Starkstein e coll. 1996). Caratteristica peculiare del DDM è la sua ciclicità: nell'arco della vita del paziente esiste un'alta probabilità che l'episodio di malattia si riproponga periodicamente, anche a distanza di tempo rispetto alla prima manifestazione. Il 93% dei soggetti che ha sperimentato un primo episodio depressivo, ne sviluppa un secondo in un periodo successivo della propria esistenza, il 60% entro 5 anni dal primo episodio. Alla luce di queste osservazioni si comprende perché la terapia del DDM necessiti di un trattamento farmacologico cronico.
Studi epidemiologici hanno dimostrato come il 40-50% dei casi di DDM sia legato a fattori ereditari: analisi su famiglie hanno evidenziato un più alto rischio di incidenza della patologia tra i parenti di primo grado di individui depressi (Gershon e coll. 1987).
Tuttavia contribuiscono alla vulnerabilità soggettiva nei confronti della depressione altri fattori non ereditari, sia endogeni che non, come lo stress, i traumi emozionali (malattie, lutti), i fattori ambientali, le infezioni virali e altri eventi dannosi verificatisi durante lo sviluppo.
L'elevata frequenza dei disturbi affettivi nei giovani sembra poi correlabile ad elementi di ordine sociale, culturale ed ambientale (disconoscimento di valori fondamentali, trasformazione dei modelli culturali, cambiamento della struttura familiare, tendenza all'individualismo e alla competizione), oltre all'uso di sostanze psicotrope e farmaci.
Secondo il DSM IV-TR, il DDM è un disturbo dell'affettività caratterizzato dalla profonda riduzione del tono dell'umore e da anedonia, cioè dalla progressiva perdita di interesse e piacere per attività normalmente gratificanti.
Oltre a questi due sintomi cardine, il DDM in fase conclamata, si manifesta tuttavia con una pletora di altri sintomi, quali tristezza, pessimismo, rallentamento dell'ideazione e dell'attività motoria, riduzione dell'autostima e della fiducia in se stessi, difficoltà a svolgere le normali attività della vita quotidiana, tendenza al pianto e ansia. Disturbi neurovegetativi frequenti sono rappresentati da alterazioni dell'appetito, con conseguente aumento o perdita di peso, e disturbi del sonno, con conseguente insonnia o ipersonnia. Nel complesso, il DDM è una condizione dominata da sentimenti spiacevoli e negativi riguardanti se stessi e il mondo circostante e dalla conseguente difficoltà a programmare il futuro. Secondo il DSM IV-TR, un paziente è affetto da DDM quando, in assenza di altre patologie, presenta tutti i giorni per almeno due settimane almeno cinque dei sintomi riportati nella Tabella 1, di cui almeno uno deve essere anedonia o umore depresso. Inoltre i sintomi devono essere tali da impedirgli di svolgere le normali attività sociali ed occupazionali.
Una forma di depressione più lieve rispetto al DDM, caratterizzata da sintomi meno gravi dal punto di vista sociale e lavorativo, è la distimia. Nell'individuo affetto da disturbo distimico i sintomi depressivi, sfumati ma costanti, possono essere considerati una caratteristica peculiare della personalità. Infatti tale patologia spesso non viene diagnosticata e, di conseguenza, il paziente non riceve un adeguato trattamento farmacologico. In molti casi, tuttavia, anche il trattamento farmacologico può non essere del tutto efficace e il paziente affetto da distimia non ritorna allo stato eutimico.
Tabella 1. Criteri per la diagnosi del Disturbo DDM
Cinque (o più) dei seguenti sintomi devono essere contemporaneamente presenti durante un periodo di 2 settimane e devono rappresentare un cambiamento rispetto al precedente livello di funzionamento; almeno uno dei sintomi è costituito da umore depresso o anedonia (perdita di interesse o piacere).
Umore depresso per la maggior parte del giorno, quasi ogni giorno, come riferito dal soggetto (per es., l'individuo si sente triste o vuoto) o come osservato dagli altri (per es., appare lamentoso). Nei bambini e negli adolescenti lo stato d'animo depresso può manifestarsi come irritabilità.
Marcata diminuzione di interesse o piacere per tutte, o quasi tutte, le attività per la maggior parte del giorno, quasi ogni giorno (come riportato dal soggetto o come osservato dagli altri).
Significativa perdita di peso, senza che il soggetto sia in regime di dieta, o aumento di peso (per es., un cambiamento superiore al 5% del peso corporeo in un mese); diminuzione o aumento dell'appetito quasi ogni giorno. Nei bambini, si osserva l'incapacità di raggiungere i normali livelli ponderali.
Insonnia o ipersonnia quasi ogni giorno.
Agitazione o rallentamento psicomotorio quasi ogni giorno (osservabile dagli altri, non semplicemente percepibile dal soggetto come irrequietezza o rallentamento)
Spossatezza, stanchezza o mancanza di energia quasi ogni giorno (rallentamento del livello di attività: il depresso si sente apatico e indolente).
Sentimenti di autosvalutazione (concetto negativo di sé), sentimenti di colpa eccessivi o inappropriati (che possono essere deliranti), quasi ogni giorno (non semplice autoaccusa o sentimenti di colpa per essere ammalato).
Ridotta capacità di pensare e di concentrarsi o indecisione quasi ogni giorno (come impressione soggettiva o osservata dagli altri).
Pensieri ricorrenti di morte (non solo paura di morire), ricorrente ideazione suicidaria senza un piano specifico, o tentativo di suicidio e ideazione di un piano specifico per commetterlo.
I sintomi devono causare un disagio clinicamente significativo o compromissione dell'interazione sociale, lavorativa o di altre aree importanti.
I sintomi non devono essere dovuti agli effetti fisiologici diretti di una sostanza (per es., una droga d' abuso, un medicamento) o di una condizione patologica generale (per es., ipotiroidismo).
Il disturbo bipolare (BD) è un disordine affettivo in cui il paziente sperimenta non solo il quadro clinico caratteristico della depressione, analogo a quello descritto per il DDM, ma anche lo stato maniacale. Negli individui affetti da BD si manifestano episodi ciclici di depressione e di mania che possono permanere da alcune settimane ad alcuni mesi.
Kandel e coll. (1994) hanno sottolineato che il 25% dei soggetti affetti da DDM possono incorrere durante l'intero arco della loro vita in almeno un episodio maniacale, ma che si possa diagnosticare il BD soltanto quando si constati l'incidenza regolare di diversi eventi depressivi e maniacali.
Anche il BD infatti è un disturbo ricorrente: dopo il primo episodio maniacale, altri episodi, sia di depressione che di mania, si succedono con una frequenza doppia rispetto alla frequenza del DDM (Kandel e coll. 1994; Goodwin e Janison, 1990).
Secondo il DSM IV-TR lo stato maniacale è caratterizzato da una sintomatologia esattamente opposta a quella dello stato depressivo. Durante la fase maniacale il soggetto affetto da BD presenta per circa una settimana in modo continuativo un tono dell'umore persistentemente elevato, risulta pervaso da un grande eccitamento, da una sensazione di esaltazione e di euforia che lo porta ad una estrema iperattività, ad una illimitata fiducia nelle proprie capacità e ad idee di grandezza. Poiché spesso tali individui possono percepire la realtà in modo distorto e poiché non sono consapevoli dei propri limiti, facilmente possono incorrere in esperienze rischiose. Inoltre la loro prorompente vitalità spesso sfocia nell'impazienza e nell'irritabilità o nella facile distraibilità da parte di stimoli esterni con conseguente diminuzione del livello di vigilanza. Anche la sfera vegetativa risulta alterata con un'aumento della libido, insonnia e iperfagia (Tabella 2).
Tra un episodio depressivo ed uno maniacale si possono presentare anche stati di umore misto o lunghi periodi di remissione durante i quali il paziente è eutimico e non presenta disturbi di alcun genere. In altri casi gli episodi depressivi e maniacali si succedono senza alcun ritorno allo stato eutimico.
Talvolta si possono riscontrare alterazioni dell'umore più lievi, come la ciclotimia, che si possono presentare anche nell'arco della stessa giornata, ma che non raggiungono l'intensità dell'episodio di depressione o di mania. Pochissimi individui soffrono solo di eventi maniacali ricorrenti, senza episodi depressivi.
Infine una piccola percentuale di pazienti bipolari, di cui l'80% sono donne, presenta un'alternanza rapida tra depressione e mania; tale rapidità di ciclo sembra correlabile con una disfunzione della tiroide. Numerosi studi clinici attribuiscono ad una scorretta terapia con antidepressivi triciclici l'insorgenza di questa rapida alternanza. L'interruzione del trattamento con triciclici e la somministrazione di composti tiroidei e sali di litio sembra consentire il ripristino della normale ciclicità (Pletscher, Shore e Brodie, 1956; Goodwin e Jamison, 1990).
Sembra che, oltre al BD a cicli rapidi, anche altri sottotipi di BD, non rispondano in modo completo alle terapie attualmente disponibili. Questi disturbi presentano perciò un alto tasso di ricadute e refrattarietà al trattamento (Coryell e coll. 1992).
E' possibile distinguere tra BD di tipo I, che si manifesta nell'1% della popolazione con i sintomi classici di mania e depressione, e BD di tipo II, che ha un'incidenza dello 0,6% nella popolazione ed è caratterizzato da episodi ipomaniacali. In quest'ultimo, meno grave rispetto al BD di tipo I, l'ipomania può anche rivelarsi come aumentata creatività del soggetto e non sempre vengono compromessi i rapporti interpersonali. L'episodio di ipomania non è associato al danneggiamento della capacità di giudizio e anzi può sfociare in un miglioramento della capacità e dell'efficienza (Bear e Canard, 2002).
Tabella 2. Criteri per la diagnosi del BD
Un periodo definito di umore anormale e persistentemente elevato, espansivo o irritabile, della durata di almeno una settimana (o di qualsiasi durata se è necessaria l'ospedalizzazione).
Durante il periodo di alterazione dell'umore, tre (o più) dei seguenti sintomi sono stati persistenti e presenti a un livello significativo (quattro se l'umore è solo irritabile):
Autostima ipertrofica o grandiosità.
Diminuito bisogno di sonno (per es., l'individuo si sente riposato dopo solo 3 ore di sonno).
Maggiore loquacità rispetto al solito, oppure spinta continua a parlare.
Fuga delle idee o esperienza soggettiva che i pensieri si succedano rapidamente.
Distraibilità (cioè l'attenzione e la vigilanza dell'individuo è troppo facilmente deviata da stimoli esterni non importanti o non pertinenti).
Aumento dell'attività finalizzata (sociale, lavorativa, scolastica o sessuale) oppure agitazione psicomotoria.
Eccessivo coinvolgimento in attività ludiche che hanno un alto potenziale di conseguenze dannose (per es., eccessi nel comprare, comportamento sessuale sconveniente, investimenti in affari avventati).
L'alterazione dell'umore è sufficientemente grave da causare una marcata compromissione dell'attività lavorativa o delle attività sociali abituali o delle relazioni interpersonali o tale da richiedere l'ospedalizzazione per prevenire danni a sé o agli altri; oppure sono presenti manifestazioni psicotiche.
I sintomi non sono dovuti agli effetti fisiologi diretti di una sostanza (per es., una droga d'abuso, un farmaco, o un altro trattamento) o ad una condizione medica patologica generale (per es., ipertiroidismo).
Nonostante il grande impatto sociale dei disturbi dell'umore, a tutt'oggi poco si conosce circa la loro eziologia e la loro patogenesi.
Negli anni '50 si osservò casualmente che la reserpina, un farmaco normalmente utilizzato per il trattamento dell'ipertensione, era in grado di indurre uno stato depressivo. Tale sostanza causa la deplezione della noradrenalina (NA) e della serotonina (5-HT) a livello sinaptico in quanto blocca lo storage di questi neurotrasmettitori nelle vescicole sinaptiche, favorendo il loro rilascio nel citoplasma, dove vengono ossidate dalle monoaminoossidasi (MAO). In conseguenza di ciò, viene ridotta la disponibilità neuronale di NA e 5-HT e, dopo alcuni giorni, si verifica l'arresto dell'attività sinaptica (Kandel e coll. 1994; Rang e coll. 2005).
Nello stesso periodo storico un altro farmaco, l'isoniazide, utilizzato per la terapia della tubercolosi, aveva dimostrato di esercitare, come effetto secondario, un miglioramento del tono dell'umore.
L'iproniazide, un analogo strutturale dell'isoniazide, fu pertanto utilizzato con successo per il trattamento della depressione nella prima metà degli anni '60. Oggi è noto che l'iproniazide è un inibitore irreversibile e non selettivo delle MAO, capace di ridurre la velocità di degradazione della NA e della 5-HT all'interno dei neuroni, accrescendone così la disponibilà sinaptica (Kandel e coll. 1994).
Lo studio del meccanismo d'azione dell'imipramina, un farmaco triciclico introdotto in terapia come antidepressivo nei primi anni '60, ha permesso di chiarire che la sua efficacia terapeutica, è determinata dalla capacità di inibire la ricaptazione neuronale di NA e 5-HT e dal conseguente potenziamento della trasmissione noradrenergica e serotoninergica (Kandel e coll. 1994; Blier e De montigny,1994).
Queste ed altre osservazioni cliniche e sperimentali hanno consentito a Schildkraut, verso la fine degli anni '60, di formulare la prima ipotesi sulla possibile patogenesi della depressione, la cosiddetta ipotesi monoaminergica. Secondo Schildkraut, la depressione potrebbe essere determinata da un deficit di NA, di 5-HT e, in misura minore, di dopamina (DA)( Sullivan e coll. 2000) a livello delle sinapsi del sistema nervoso centrale (SNC). Tale ipotesi è suffragata dall'osservazione che le prime classi di farmaci efficaci nel trattamento della depressione, gli antidepressivi triciclici e gli inibitori delle monoaminoossidasi, facilitano e potenziano la neurotrasmissione monoaminergica, soprattutto noradrenergica e serotoninergica. Tali farmaci infatti aumentano la concentrazione di NA e 5-HT a livello sinaptico, i triciclici attraverso l'inibizione della ricaptazione neuronale delle monoamine e gli inibitori delle MAO attraverso il blocco della loro degradazione.
I sistemi monoaminergici hanno una distribuzione diffusa in tutto il SNC, in particolare in alcune aree, quali la corteccia frontale, lo striato e il sistema limbico. Tali aree presentano un elevato livello di integrazione e di interconnessioni reciproche e formano un network in grado di regolare l'emotività, la sfera cognitiva ed affettiva e di coordinare le risposte motorie, comportamentali e vegetative ad esse correlate.
Le vie serotoninergiche, che originano nei nuclei del rafe, regolano diverse funzioni quali la memoria, lo stato dell'umore, il sonno, l'appetito, il vomito, l'attività sessuale, la temperatura corporea.
La maggior parte dei neuroni noradrenergici origina invece dal locus coeruleus e regola le funzioni cognitive, le risposte emotive, il tono dell'umore, l'attenzione, la motivazione e, attraverso il sistema ortosimpatico, le funzioni cardiovascolari e della gran parte degli organi interni.
Negli anni passati, allo scopo di verificare l'ipotesi monoaminergica, sono stati effettuati numerosi studi per valutare le modificazioni della concentrazione di NA, di 5-HT e dei loro metaboliti nei liquidi biologici (fluido cerebrospinale, sangue, urine) e nei tessuti cerebrali dei pazienti affetti da depressione o mania e per valutare l'efficacia dei precursori della 5-HT nella terapia della depressione.
Il principale metabolita centrale della NA è l'MHPG (3-metossi-4-idrossi-feniletilenglicole), la cui concentrazione è stata studiata per lo più nell'urina, in quanto i suoi livelli liquorali sembrano rappresentare un indice del metabolismo noradrenergico spinale più che cerebrale. Evidenze sperimentali suggeriscono che il 20-80% dell'MHPG eliminato con l'urina deriva dal metabolismo della NA a livello cerebrale. Rispetto ai soggetti normali, nei pazienti bipolari si sono riscontrati livelli medi di MHPG significativamente ridotti durante la fase depressiva e lievemente ridotti nella fase maniacale. Nei pazienti depressi unipolari sono stati invece rilevati livelli di MHPG ridotti, più elevati o uguali rispetto ai controlli. Probabilmente i livelli più elevati di NA rilevati nei pazienti depressi rispetto ai soggetti normali riflettono l'aumento dell'attività simpatica periferica causata dall'ansia, spesso associata alla depressione (Paoletti e coll, 1999). Livelli urinari ridotti di MHPG sono stati riscontrati in pazienti unipolari durante la fase acuta della malattia, ma negli stessi pazienti le concentrazioni di MHPG tendono a normalizzarsi con la risoluzione dell'episodio depressivo. Questo dato ha portato ad ipotizzare che i livelli urinari di MHPG riflettano alterazioni transitorie e contingenti legate alla fase della malattia piuttosto che alla presenza della patologia stessa. Inoltre, alcuni autori sostengono che l'escrezione urinaria di questo metabolita sia influenzata da una serie di variabili aspecifiche, come l'ttività motoria, il regime alimentare e il ciclo sonno-veglia. Pertanto la valutazione della concentrazione urinaria di MHPG non sembra avere valore predittivo e non avvalora l'ipotesi monoaminergica.
Il principale metabolita della 5-HT a livello cerebrale è il 5-HIAA (acido 5-idrossi-indolacetico). Anche i dati relativi alle concentrazioni di 5-HIAA nei liquor e nell'urina dei pazienti depressi appaiono discordanti. Alcuni studi hanno riportato livelli ridotti del metabolita in circa il 30-40% dei pazienti depressi, mentre in altri non è stata rilevata alcuna significativa differenza rispetto ai soggetti di controllo. Altri studi hanno suggerito una probabile correlazione tra bassi livelli di 5-HIAA e comportamento suicidario (maggiore frequenza di tentativi di suicidio) (Mann e coll. 1999). Gli autori, partendo dal presupposto che i bassi livelli di 5-HIAA fossero una conseguenza della carenza di 5-HT cerebrale, si sarebbero aspettati un aumento dei livelli del metabolita in seguito al trattamento con antidepressivi, almeno nei pazienti rispondenti alla terapia, ma i risultati delle analisi hanno fornito dati contrastanti e spesso di difficile interpretazione. Nel loro complesso questi studi non hanno fornito dati sufficienti a dimostrare una correlazione tra concentrazioni di 5-HIAA nei liquidi biologici e il DDM.
Per quanto concerne gli studi clinici sull'efficacia antidepressiva di precursori della serotonina come il L-triptofano (TRP) e il 5-idrossi-triptofano (5-HTP), tali precursori non hanno prodotto risultati significativi e l'efficacia di questi farmaci risulta essere per lo più modesta.
La stessa osservazione "storica" sulle proprietà depressive della reserpina è stata recentemente riconsiderata. Goodwin e coll. (1990) hanno osservato che solo una piccola percentuale di pazienti (5-10%) trattati con reserpina sviluppano una vera e propria sintomatologia depressiva, mentre la maggior parte presenta una condizione "pseudo-depressiva" caratterizzata soprattutto da sedazione e letargia.
E' stato infine sottolineato che l'inibizione della ricaptazione delle monoamine cerebrali, con il conseguente potenziamento della trasmissione monoaminergica, debba essere considerato come il meccanismo iniziale (acuto) dell'azione antidepressiva e che non possa di per sé mediare gli effetti terapeutici di questi farmaci. Infatti la risposta terapeutica si manifesta dopo alcune settimane rispetto all'inizio della terapia. Livelli plasmatici di antidepressivo efficaci nel produrre il blocco della ricaptazione e il potenziamento monoaminergico si raggiungono dopo poche ore, in altre parole gli effetti del farmaco sui principali bersagli molecolari sono osservabili in tempi brevi, ma l'effetto terapeutico nei confronti dei sintomi depressivi compare dopo almeno 4-6 settimane di trattamento. Per spiegare questo periodo di latenza nella comparsa dell'effetto antidepressivo, è stata avanzata l'ipotesi che la somministrazione cronica del farmaco inneschi risposte adattative da parte dell'organismo e che siano questi meccanismi omeostatici i veri responsabili dell'efficacia terapeutica.
D'altra parte non esistono evidenze dirette che la principale causa della depressione siano le anomalie dei meccanismi che controllano la neurotrasmissione monoaminergica. Gli studi in parte sopra descritti non hanno definito con chiarezza il contributo di tali sistemi neurochimici all' eziopatogenesi della depressione. Quindi, per quanto la disregolazione dei sistemi monoaminergici sia certamente coinvolta nei disturbi dell'umore, essa non è sicuramente l'elemento scatenante che determina l'insorgenza della patologia.
Inoltre non tutti i farmaci che sono in grado di aumentare la neurotrasmissione noradrenergica e serotoninergica, come per esempio gli psicostimolanti, sono farmaci antidepressivi.
Al contrario non tutti i farmaci dotati di attività antidepressiva sono in grado di inibire significativamente la ricaptazione sinaptica delle monoamine, come ad esempio l'iprindolo.
Per quanto sia evidente la compromissione dei sistemi monoaminergici nella depressione, un semplice "modello sinaptico" non può spiegare in modo esaustivo l'eziopatogenesi della malattia.
I più recenti sviluppi della ricerca nel campo dei disturbi affettivi hanno consentito di sviluppare altre ipotesi sui possibili meccanismi cellulari che sottendono la fisiopatologia della depressione, non necessariamente in contraddizione con la precedente ipotesi monoaminergica.
Tali ipotesi offrono anche una convincente interpretazione del meccanismo d'azione dei farmaci antidepressivi. Questi composti agirebbero modificando gli eventi regolatori intraneuronali, con una sequenzialità temporale sovrapponibile alla latenza necessaria per la comparsa dell'effetto terapeutico. Gli antidepressivi non sarebbero soltanto in grado di interferire con la ricaptazione delle monoamine, ma anche con i meccanismi di trasduzione del segnale promossi da questi stessi neurotrasmettitori, innescando processi adattativi specifici nei pathways di segnalazione intracellulare (Manji e coll. 2000; Manji e coll. 2000).
Tali eventi comprendono i cambiamenti dell'espressione e dell'attività di recettori accoppiati a proteine G, l'attivazione di protein-chinasi specifiche, le modificazioni dell'espressione genica di fattori di trascrizione e di fattori neurotrofici.
Recenti studi hanno dimostrato che l'eccessiva esposizione a situazioni di stress può rappresentare un fattore scatenante per l'insorgenza della depressione. Circa il 50% dei pazienti affetti da DDM presenta iperattività dell'asse ipotalamo-ipofisi-surrene (HPA) e Lanfuney e coll. (2000) hanno sottolineato rilevanti e reciproche interazioni tra il sistema serotoninergico e la funzionalità dell'asse HPA nella depressione. Inoltre lo stress cronico può indurre una riduzione della sintesi di fattori neurotrofici: tali proteine sono importanti per il corretto funzionamento neuronale in quanto non solo hanno un ruolo fondamentale durante lo sviluppo del SNC, ma sono determinanti nei fenomeni di plasticità sinaptica.
E' stato infatti suggerito che i disturbi dell'umore sono associati ad alterazioni della plasticità neuronale (Manij e coll. 2000; Manij e coll. 2000) e che i pathways di segnale coinvolti nella sopravvivenza e nella morte cellulare siano i bersagli a lungo termine dell'azione farmacologica. Gli antidepressivi e il litio, un farmaco stabilizzante dell'umore, modulano indirettamente un certo numero di fattori coinvolti nei meccanismi di sopravvivenza e morte cellulare, tra cui la proteina CREB (Cyclic AMP Responsive Element Binding Protein) e la proteina BDNF (Brain-derived Neurotropic Factor) (Duman e coll. 1997).
Duman e coll. (1997) hanno inoltre dimostrato che farmaci antidepressivi, anche molto diversi dal punto di vista chimico, possiedono in comune la capacità di aumentare l'espressione di proteine ad azione neuroprotettiva.
Il potenziamento sinaptico della trasmissione monoaminergica indotto dagli antidepressivi attraverso l'inibizione delle MAO o il blocco della ricaptazione o la down-regolazione dell'autorecettore presinaptico/somatodendritico, determinerebbe quindi una maggiore attivazione dei recettori post-sinaptici, accoppiati a meccanismi di trasduzione del segnale. La ripetuta attivazione di queste cascate intracellulari porterebbe alla attivazione di fattori di trascrizione che, a loro volta, sarebbero in grado di controllare l'espressione di geni coinvolti nella sopravvivenza delle cellule nervose.
Duman e coll. (1997) in seguito a terapia cronica con antidepressivi hanno osservato nell'ippocampo di ratto un aumento della concentrazione del mRNA che codifica per il fattore di trascrizione CREB e un aumento della concentrazione della proteina stessa. Sulla base di tale evidenza hanno ipotizzato che CREB sia responsabile dell'attivazione dei geni che controllano l'espressione del BDNF e del suo recettore Trk.
Il BDNF è una neurotrofina appartenente ad una famiglia di fattori di crescita deputati al controllo di numerose attività neuronali, quali i meccanismi di differenziazione durante lo sviluppo e quelli di sopravvivenza nel cervello adulto.
In accordo con l'ipotesi sopra formulata, gli autori hanno osservato un aumento dei livelli del BDNF e del suo mRNA nell'ippocampo dei ratti trattati in modo cronico con un' ampia varietà di antidepressivi.
A conferma di ciò, è stato dimostrato che alcuni pazienti depressi presentano livelli sierici di BDNF più bassi rispetto a soggetti sani di controllo (Smith e coll. 1995). Gli stessi autori hanno dimostrato che lo stress può ridurre l'espressione del BDNF nell'ippocampo del ratto e che tale effetto può essere contrastato dal trattamento cronico (ma non acuto) con farmaci antidepressivi. Infine, Siuciak e coll. (1997) hanno riportato che l'infusione intracerebrale di BDNF ha effetti antidepressivo-simili.
Sulla base di queste e numerose altre evidenze sperimentali oggi si ritiene che i disturbi affettivi possano essere provocati da fattori individuali (genetici) e da fattori legati all'ambiente e alle condizioni di vita, che interverrebbero durante lo sviluppo o nel corso della vita adulta, contribuendo in misura diversa al deterioramento della capacità omeostatica delle cellule e alla progressiva perdita della plasticità neuronale. Le neurotrofine, tra cui il BDNF, favoriscono la sopravvivenza della cellula attivando diverse vie di propagazione del segnale, tra cui il pathway della PI-3-Kinasi e quello della MAP-Kinasi. La cascata della MAP-Kinasi prevede la fosforilazione di intermedi quali Ras, Raf, MEK ed ERK. Uno dei bersagli di ERK è RSK che può influenzare la sopravvivenza cellulare in due modi: 1) inattivando mediante fosforilazione il fattore pro-apoptotico BAD, aumentando così la trascrizione del gene anti-apoptotico Bcl2 e 2) fosforilando CREB, che a sua volta aumenta la trascrizione del gene BDNF.
Oltre che dalla via della MAP-Kinasi (Ghosh e coll. 1994), CREB può essere attivato mediante fosforilazione da parte della PKA, attivata dei recettori β-adrenergici (Roseboom e Klein, 1995) e della PKC, attivata dai recettori α1-adrenergici e 5-HT2 serotoninergici (Duman, 1998).
La ricerca preclinica ha chiaramente evidenziato che i farmaci antidepressivi sono in grado di promuovere la fosforilazione di CREB e, di conseguenza, aumentare l' espressione del BDNF.
Dati post-mortem evidenziano che pazienti depressi non trattati farmacologicamente presentano livelli di CREB inferiori nella corteccia frontale e nell'ippocampo rispetto ai soggetti sani.
Uno dei principali meccanismi attraverso cui il BDNF promuove la sopravvivenza della cellula è l'aumento dell'espressione delle principali proteine anti-apoptotiche (quindi citoprotettive), Bcl-2 e Bcl-X, rispetto a BAD e BAX, altri membri pro-apoptotici della famiglia Bcl. E' noto che il mantenimento dell'equilibrio tra i livelli dei fattori pro- ed anti-apoptotici può influenzare la sopravvivenza delle cellule neuronali nel SNC.
Bcl-2 controlla il processo di morte cellulare programmata attraverso la regolazione del rilascio di Ca++ e del Citocromo C dai mitocondri. Il Citocromo C nel citoplasma della cellula è in grado di sequestrare la preforma degli enzimi induttori di morte, le caspasi, interrompendo il segnale intracellulare di morte.
Il litio e il valproato, due farmaci stabilizzanti dell'umore, hanno effetti neuroprotettivi in quanto aumentano l'espressione di Bcl-2 e inibiscono la kinasi GSK-3β. Il valproato sembra anche in grado di attivare il pathway della MAP-Kinasi (Manij e coll. 2000; Manij e coll. 2000).
Recenti studi hanno rivelato che gli antidepressivi aumentano l'espressione non solo di CREB fosforilato (pCREB), ma anche dei geni che codificano per CAM-L1, proteina L1 di adesione cellulare, e per la laminina (Laifenfeld e coll. 2002a, Laifenfeld e coll. 2002b; Laifenfeld e coll. 2005). Tutte queste molecole sembrano coinvolte nei processi di crescita, sopravvivenza e differenziamento neuronale (Hall e coll. 1997, Hall e coll. 2000; Kamiguchi e Lemmon, 1997; Karin e Hunter, 1995; Luthl e coll. 1994). Sembra che per attivare i fenomeni di plasticità sia necessario che CREB leghi Barx2 e che tale legame influenzi la trascrizione di CAM-L1 (Crossin e Krushel, 2000); a ciò pare che debba seguire il legame di CAM-L1 alla Laminina (Hall e coll. 1997, Hall e coll. 2000).

Figura 1. Ipotesi sulla neuropatogenesi della depressione
La plasticità sinaptica è un meccanismo dinamico che consente alle reti neuronali di adattare e modellare la propria struttura e e le proprie funzioni in risposta agli stimoli interni ed esterni (Zilles e coll. 1992).
Questa capacità di modificare l'efficacia della connettività sinaptica è una caratteristica determinante per l'adattamento e la sopravvivenza degli organismi viventi. Nel cervello, in particolare nel sistema limbico e nella corteccia frontale, tali modificazioni della connettività sono alla base dell'apprendimento e della memoria, ma determinano anche le risposte allo stress e i sintomi cognitivi, l'ansia e i disturbi della sfera emozionale che accompagnano la depressione (Le Doux, 1996; Sweatt, 2001).
Alcuni studi in vivo hanno mostrato che la terapia elettroconvulsiva (ECS) aumenta la plasticità neuronale nel giro dentato dell'ippocampo del piccolo roditore (Steward and Reid, 1993; Reid and Steward, 1997). Gli stessi autori hanno riportato che anche la somministrazione di fluoxetina, un antidepressivo che blocca in modo selettivo la ricaptazione della 5-HT, provoca lo stesso effetto. Queste evidenze sono coerenti con le osservazioni cliniche riguardanti l'efficacia della ECT nel trattamento del DDM (Steward and Reid, 1993; Steward and Reid, 2000; Reid and Steward, 1997; Reid and Steward, 2001).
Petrie e coll. (2000) hanno suggerito che la capacità di potenziare la plasticità sinaptica (e non solo la capacità di favorire la sopravvivenza neuronale) sia la proprietà che determina l'efficacia terapeutica dei farmaci antidepressivi.
Numerose evidenze hanno confermato che, in soggetti predisposti, lo stress è associato al rischio di insorgenza di disturbi dell'umore.
L'aumento dei livelli endogeni di glucocorticoidi che si verifica in seguito a stress acuto ha effetti importanti sull'ippocampo e sui meccanismi che regolano la plasticità neuronale. Nell'ippocampo sono presenti recettori per i corticosteroidi, sia di tipo I ad alta affinità, sia di tipo II a bassa affinità.
L'attivazione di uno o l'altro di questi sottotipi recettoriali da parte del corticosterone upregola o downregola la connettività sinaptica in modo concentrazione-dipendente (Pavlides e coll. 1995). Alti livelli di corticosterone, il principale ormone dello stress nei roditori, riducono la capacità dei neuroni ippocampali di creare nuovi contatti sinaptici. Al contrario, bassi livelli di corticosterone, che attivano il recettore di tipo I, aumentano la plasticità sinaptica. Agonisti selettivi per il recettore di tipo II mimano gli effetti di alti livelli di corticosterone, mentre composti antagonisti del recettore di tipo II contrastano le modificazioni indotte dallo stress sulla plasticità sinaptica (Xu e coll, 1998).
Gli effetti dei corticosteroidi potrebbero essere legati all'attività del BDNF, in quanto alti livelli di corticosterone downregolano la trascrizione del gene BDNF e riducono i livelli della neurotrofina nell'ippocampo (Schaaf e coll, 1998).
La rimozione delle ghiandole surrenali previene la diminuzione stress-indotta della plasticità neuronale, ma il fenomeno è aumentato in seguito a somministrazione di glucocorticoidi.
Studi sulla relazione tra stress e plasticità neuronale hanno consolidato l'opinione che eventi avversi ed esperienze negative sperimentati nell'età dello sviluppo possano accrescere la vulnerabilità del soggetto e favorire la comparsa di disturbi dell'umore in età più avanzata. Kehoe e coll. (1995) hanno dimostrato nel giro dentato dell'ippocampo di ratti neonati, che hanno sperimentato episodi ripetuti di breve separazione dalla madre (un'ora al giorno per otto giorni consecutivi), un' aumento dell'induzione e della durata dell'LTP (Long-Term Potentiation); dopo tre settimane compaiono evidenti modificazioni della plasticità neuronale con effetti maggiori nel maschio rispetto alla femmina.
Alcuni autori hanno infine osservato che l'esposizione ad alcuni tipi di stress nell'infanzia porta ad alterazioni persistenti dell'asse HPA, dei sistemi serotoninergico e noradrenergico centrali e del sistema nervoso simpatico, modificazioni che possono determinare una più accentuata vulnerabilità soggettiva nei confronti di stress conseguenti.
I modelli animali costituiscono strumenti indispensabili per la ricerca in campo biologico in quanto consentono di riprodurre in organismi più semplici le caratteristiche essenziali di alcune patologie umane e, di conseguenza, di individuare i possibili bersagli per la messa a punto di nuove strategie terapeutiche (Suomi, 1982; McKinney, 1988).
I modelli animali dei disturbi psichiatrici possono essere definiti operativamente come "insoliti stati comportamentali" che possono essere contrastati in modo specifico dalla stessa terapia farmacologica che annulla i sintomi della malattia nell'uomo (Petty e Sherman, 1981). Un modello animale di disturbo psichiatrico idealmente dovrebbe essere semplice, riproducibile, e presentare analogie con il disturbo umano nella sintomatologia, nell'eziopatogenesi e nella risposta al trattamento (Murphy, 1977; Crawley e coll. 1984; Crawley e coll. 1985; Willner, 1984). La depressione, come molte patologie psichiatriche, coinvolge processi cognitivi, emozionali e motivazionali difficilmente riproducibili nell'animale da laboratorio. Allo stato attuale infatti non esistono modelli animali di depressione che soddisfano in pieno i requisiti essenziali precedentemente esposti. Il problema diventa più complesso se si prende in considerazione il BD, nel quale si alternano episodi ricorrenti di mania e depressione. Il limite principale per la messa a punto di un modello animale di BD è rappresentato proprio dall' impossibilità, al momento, di riprodurre la ciclicità dell'umore.
Prendendo in considerazione il DDM, il numero e l'ampia varietà di sintomi che caratterizzano il disturbo rappresentano l'ostacolo maggiore alla realizzazione di un modello preclinico. Inoltre, alcuni sintomi del DDM, come l' ideazione suicidaria, la ridotta autostima e i ricorrenti sensi di colpa, non possono essere ricreati negli animali.
Nonostante le evidenti difficoltà, negli ultimi decenni sono stati sviluppati modelli preclinici di depressione che riproducono in modo convincente, sia nel ratto che nel topo, alcuni sintomi cardine della depressione umana. L'uso di tali modelli animali può fornire importanti informazioni sui meccanismi fisiopatologici della depressione e, allo stesso tempo, è utile per individuare i possibili bersagli per nuovi farmaci antidepressivi e per valutarne l'efficacia terapeutica prima della sperimentazione sull'uomo.
Circa trenta anni fa McKinney e Bunney hanno suggerito i requisiti essenziali per un buon modello preclinico di depressione:
il modello deve essere "ragionevolmente analogo" alla patologia umana per ciò che concerne la sintomatologia;
il modello deve produrre nella specie animale utilizzata modificazioni del comportamento che possano essere oggettivamente misurate e monitorate;
le modificazioni comportamentali dovrebbero essere prevenute e contrastate dalle stesse strategie terapeutiche efficaci nel trattamento della depressione umana (il modello cioè deve rispondere adeguatamente ai farmaci antidepressivi);
il modello animale deve essere facilmente riproducibile.
In tempi più recenti, Willner e coll. (1991) hanno elaborato ulteriormente queste problematiche, fissando i criteri di validità di un modello animale di depressione:
la "validità predittiva" o "isomorfismo farmacologico" è definita come la capacità del modello animale di rispondere ai farmaci antidepressivi, in altre parole si deve poter verificare la corrispondenza tra gli effetti dei farmaci nel modello e in clinica;
la "validità d'aspetto" è la corrispondenza tra le modificazioni comportamentali prodotte nel modello animale e i sintomi della patologia umana;
la "validità del costrutto" è la corrispondenza tra il modello e la patologia umana alla luce delle attuali conoscenze sull'eziopatofisiologia della depressione.
Molti modelli preclinici di depressione attualmente disponibili hanno una buona validità predittiva, con relativamente pochi falsi positivi e falsi negativi, pochi modelli rispecchiano più di due criteri di validità e, infine, soltanto alcuni modelli sono coerenti con la cronicità della depressione umana (Willner 1984, Willner 1990).
E' chiaro che la validità di ogni modello preclinico, secondo i criteri precedentemente citati, deve essere valutata con estrema cautela prima di estrapolare e trasferire i risultati ottenuti dall'animale all'uomo (Yadid, 1998; Cryan, Markou e Lucki, 2002).
I modelli di depressione oggi in uso sono stati sviluppati utilizzando le conseguenze dello stress, somministrato durante lo sviluppo o in età adulta, le modificazioni indotte da intervento chirurgico/trattamento farmacologico o la selezione fenotipica.
La rimozione bilaterale dei bulbi olfattivi nel ratto, nel topo e nel criceto causa una complessa varietà di alterazioni comportamentali, neurochimiche, endocrine e del sistema immunitario, molte delle quali correlabili con i sintomi osservati nel DDM (Kelly e coll. 1997).
La più rilevante modificazione del comportamento provocata dalla bulbectomia olfattiva è rappresentata da un aumento dell'attività locomotoria, normalizzato dal trattamento cronico, ma non acuto, con farmaci antidepressivi, (Kelly e coll. 1997; Cryan e coll. 1998).
Recenti studi hanno suggerito che l'iperattività motoria sia correlabile ad una esaltazione del comportamento difensivo (Stock e coll. 2001) o ad alterazioni del comportamento avversivo (Primeaux e Holmes, 1999).
Mar e coll. (2000) hanno anche riportato che il trattamento con farmaci antidepressivi aumenta nell'animale bulbectomizzato la capacità di abituarsi alla novità e di adattarsi a stimoli ambientali di varia natura e che tali effetti non sono secondari all'anosmia (perdita dell'odorato). Altri autori hanno focalizzato l'attenzione sulle alterazioni neurochimiche e molecolari che sottendono le modificazioni comportamentali sensibili al trattamento con antidepressivi.
Sono associate alla bulbectomia olfattiva modificazioni della funzionalità dei sistemi serotoninergico, colinergico, GABAergico, noradrenergico, glutamatergico e un aumento dell'escrezione notturna del corticosterone, soppressa dal dexametazone (Kelly e coll. 1997).
Connor e coll. (1999) sostengono invece che l'animale bulbectomizzato rappresenti un modello di depressione caratterizzata da ipofunzionalità serotoninergica, in quanto sono state osservate modificazioni dell'innervazione da parte della 5-HT nella corteccia frontale (Zhou e coll. 1998) e un aumento delle risposte stress-indotte da parte del sistema serotoninergico (Connor e coll. 1999).
Inoltre è stato dimostrato un aumento del rilascio del glutammato nello striato correlabile con l'iperattività motoria dell'animale, il quale potrebbe avere un ruolo modulatorio nella risposta agli antidepressivi (Ho e coll. 2000). Altri autori hanno rilevato aumenti della concentrazione di alcuni neuropeptidi, come il CRH, il TRH, la somatostatina (Bissette, 2001) e il neuropeptide Y (Holmes e coll. 1998), che potrebbero anch'essi giocare un ruolo nella comparsa delle modificazioni del comportamento peculiari del modello.
Immagini di risonanza magnetica hanno mostrato alterazioni d'intensità del segnale nella corteccia, nell'ippocampo, nel caudato e nell'amigdala negli animali sottoposti ad asportazione dei bulbi olfattivi rispetto ad animali di controllo (Wrynn e coll. 2000). Negli animali bulbectomizzati è evidente anche un allargamenti dei ventricoli (Wrynn e coll. 2000), fenomeno che gli autori hanno correlato con i cambiamenti morfologici osservati in alcuni pazienti depressi.
Fu sviluppato da Porsolt e coll. nel ratto (Porsolt, 1997) e, successivamente, nel topo (Porsolt, 2000). E' il modello preclinico più utilizzato per valutare l'efficacia dei farmaci antidepressivi. La larga diffusione di tale modello è dovuta alla buona validità predittiva e alla relativa semplicità del protocollo (Borsini e Mell, 1998). Il test si basa sull'osservazione che l'animale, se posto all'interno di un cilindro pieno d'acqua dal quale non può uscire, inizialmente si muove concitatamente tentando di fuggire ma, dopo un certo tempo, vista l'impossibilità di fuga, tende a restare immobile, galleggiando sul pelo dell'acqua. Se, dopo 24 ore, l'animale viene posto nuovamente nel cilindro, assume una postura immobile per quasi tutta la durata del test. Si ritiene che l'immobilità rifletta la rinuncia da parte dell'animale a mantenere un comportamento orientato alla fuga (Lucki, 1997): l'animale infatti sviluppa un comportamento passivo mirato ad evitare lo stress derivante dalla ricerca di una via di salvezza (Lucki, 1997). Se tra le due sessioni sperimentali del test di Porsolt l'animale viene trattato in acuto con farmaci antidepressivi, il tempo di immobilità si riduce. Quasi tutti i farmaci antidepressivi oggi in uso riducono in modo significativo rispetto ai controlli il tempo di immobilità dell'animale nel corso della seconda sessione e ne aumentano di conseguenza il comportamento orientato alla fuga.
La debolezza di questo modello preclinico di depressione risiede nella constatazione che l'efficacia antidepressiva compare dopo somministrazione acuta (quindi con un andamento temporale diverso da quello clinico) e nella necessità di somministrare dosi ben più elevate di quelle utilizzate in terapia umana, per esempio per valutare l'efficacia degli inibitori selettivi della ricaptazione della 5-HT (SSRIs) (Lucki, 1997).
Nel tentativo di porre rimedio a
queste limitazioni del test di Porsolt tradizionale, sono state apportate alcune
modifiche al protocollo sperimentale (Lucki, 1997). Tali variazioni comprendono
l'aumento della profondità dell'acqua nel cilindro (
Il test di Porsolt così modificato ha una maggiore validità predittiva e consente di acquisire utili informazioni sul possibile meccanismo d'azione del farmaco in esame (Lucki, 1997; Cryan e Lucki, 2000). Infatti, i farmaci antidepressivi con meccanismo noradrenergico, come la reboxetina, causano una diminuzione del tempo d'immobilità dell'animale e un aumento del tempo di climbing, mentre gli antidepressivi con meccanismo serotoninergico riducono il tempo d'immobilità ma aumentano il tempo di swimming.
Il modello di learned helplessness, ideato da Seligman e Beagley (1975) nella prima metà degli anni '70, è concettualmente simile al test di Porsolt e rappresenta uno tra i più utilizzati modelli preclinici di depressione.
Si basa sull'osservazione che gli animali, sottoposti ad una serie di eventi stressanti ai quali non possono sottrarsi, sviluppano un deficit nel comportamento di evitamento. Il protocollo è strutturato in due fasi successive. Nella prima fase l'animale viene sottoposto ad una serie di stimoli dolorosi (scossa nelle zampe attraverso il pavimento elettrificato della gabbia) somministrati in modo casuale per 3-5 giorni consecutivi. La somministrazione di stimoli dolorosi che non è possibile evitare, causa nell'animale uno stato anedonico che si protrae per circa una settimana. Oltre all'anedonia, rilevabile con la diminuzione della preferenza al saccarosio, negli animali sottoposti alla fase di pretrattamento compaiono perdita di peso, iperattività motoria, disturbi del sonno e alterazioni dell'asse HPA (Weiss, 1968, Wagner e coll. 1977). Quando nella seconda fase l'animale viene sottoposto ad un test di evitamento passivo, la performance risulta significativamente diversa rispetto agli animali di controllo, in quanto, negli animali stressati, aumenta la frequenza di scosse non evitate (Rosellini e De Cola, 1981). I farmaci antidepressivi (TCA, SSRI, iMAO, mianserina) (Weiss e coll. 1998) sono efficaci nel migliorare la performance negli animali stressati; anche un trattamento non farmacologico come uno shock elettroconvulsivo è efficace in questo modello preclinico di depressione.
Il modello animale di stress cronico variato (CMS), ideato da Katz (1982) e perfezionato successivamente da Willner (1984, 1997), è basato sull' induzione di uno stato anedonico nel ratto (Willner e coll. 1987; Willner e coll. 1992) o nel topo (Montleon e coll. 1994), mediante l'esposizione a blandi stimoli stressogeni di natura diversa, somministrati in modo non prevedibile per un periodo di alcune settimane.
L'anedonia è misurata con tecniche non invasive quali, per esempio, il progressivo calo della naturale preferenza dell'animale al saccarosio.
Gli stimoli previsti dal protocollo originale sono la privazione di cibo o di acqua, l'inversione del ciclo luce-buio, l'inclinazione o lo scuotimento della gabbia, l'affollamento della gabbia, il nuoto forzato in acqua fredda, la somministrazione di scosse elettriche nelle zampe e lo stress da costrizione. Willner e coll. (1987) hanno successivamente modificato il protocollo, escludendo gli stimoli troppo intensi ed introducendo invece stimoli stressogeni più blandi per un tempo più lungo. In queste condizioni lo stato anedonico si mantiene per alcune settimane, riproducendo l'aspetto di cronicità della patologia umana.
Oltre alla diminuzione delle risposte agli stimoli appetitivi, il CMS provoca altri sintomi paragonabili a quelli della depressione umana. Sono stati osservati, da numerosi ricercatori, diminuzione del comportamento sessuale e del comportamento esplorativo, aggressività e rallentamentio dell'attività motoria (D'Aquila e coll. 1994). Gli animali rispondono allo stress cronico anche con cambiamenti nei ritmi circadiani (Gorka e coll. 1996) e con disturbi del sonno (Moreau e coll. 1995; Cheeta e coll. 1997). Inoltre mostrano un'aumentata attività dell'asse HPA, ipertrofia cortico-surrenale (Muscat e Willner, 1992) e ipersecrezione di corticosterone (Ayensu e coll. 1995). Sono state descritte anche alterazioni del sistema immunitario con aumento dei livelli di proteine del sistema del complemento e di proteine caratteristiche della fase acuta dell'infiammazione (Ayensu e coll. 1995), diminuzioni del peso del timo, minore attività dei linfociti natural killer e della reattività agli stimoli mitogeni da parte dei linfociti T (Kubera e coll. 1994, Kubera e coll. 1995).
L'anedonia, definita come la diminuzione della capacità di sperimentare piacere (Fawcett e coll. 1983), nel modello CMS è rappresentata soprattutto da una diminuzione della sensibilità alla ricompensa. Nel protocollo sperimentale CMS lo stato anedonico viene misurato in base alla diminuzione della preferenza nei confronti di una soluzione di saccarosio (1-2%), normalmente molto gradita all'animale.
Il modello, dunque, è basato su due presupposti, vale a dire che la soluzione di saccarosio rappresenti una valida misura della soglia di autogratificazione e che questa sia aumentata, e non diminuita, dallo stress cronico.
Numerose evidenze sperimentali hanno dimostrato che durante il CMS non si misura una diminuzione del consumo di acqua pura (Muscat e Willner, 1992), dunque il decremento dell'assunzione della bevanda al saccarosio non può essere spiegato semplicemente con una variazione del consumo di liquidi. Inoltre le calorie contenute nel saccarosio sono risultate ininfluenti, in quanto effetti simili sono stati osservati in animali che consumano soluzioni di saccarosio ipocaloriche (Willner e coll. 1987; Ayensu e coll. 1995), e una diminuzione del consumo di soluzione al saccarosio è stata notata anche gli animali preventivamente a digiuno (Muscat e Willner, 1992). Anche il consumo di cibo non subisce variazioni durante il CMS, mentre sono modificate le proprietà gratificanti dell'alimento, come indicato dallo scarso interesse per il luogo di consumo del pasto (Papp e coll. 1991; Muscat e coll. 1992; Willner e coll. 1994) e da un'accelerazione dell'ingestione dello stesso (Sampson e coll. 1991). Infine, l'osservazione che il CMS induca anedonia non è basata esclusivamente sui dati sperimentali che misurano la preferenza al saccarosio, ma è supportata anche dalla verifica che il CMS causa un aumento della soglia di autostimolazione cerebrale. Lo stress cronico infatti causa un aumento della corrente-soglia necessaria per mantenere l'autostimolazione intracranica sugli elettrodi impiantati nell'area tegmentale ventrale del mesencefalo (Moreau e coll. 1992; Moreau e coll. 1993; Moreau e coll. 1994a; Moreau e coll. 1994b; Moreau e coll. 1995).
Come nell'uomo, la terapia cronica con farmaci antidepressivi è in grado di annullare i sintomi provocati dal CMS con una latenza di 3-4 settimane.
Gli antidepressivi considerati efficaci per il trattamento dell'anedonia provocata da CMS sono: i triciclici imipramina, desmipramina, amitriptilina (Willner e coll. 1987; Muscat e coll. 1990; Papp e coll. 1996; Sluzewska e Szczawinska, 1996a; Valverde e coll. 1997), gli SSRI fluoxetina, fluvoxamina, citalopram (Muscat e coll. 1992; Przegalinski e coll. 1995; Marona-Lewicka e Nichols, 1996; Sluzewska e Szczawinska, 1996a, Sluzewska e Szczawinska, 1996b), la maprotilina uno specifico inibitore della ricaptazione del Na (Muscat e coll. 1992), il moclobemide un iMAO (Moreau e coll. 1993), il brofaromine (Papp e coll. 1996) e la mianserina un antidepressivo atipico (Cheeta e coll. 1994; Moreau e coll. 1994a). In tutti gli studi, i sopra citati farmaci, sono efficaci a dosi moderate (in molti casi comprese nel range terapeutico) e, come in terapia umana, una completa risposta al trattamento richiede tipicamente 3-5 settimane. Altri farmaci, meno convenzionali, ma ugualmente efficaci nel modello CMS includono gli stabilizzanti dell'umore litio (Sluzewska e Szczawinska, 1996a) e carbamazepina (Sluzewsaka e Nowakowska, 1994), il farmaco ansiolitico buspirone (Przegalinski e coll. 1995; Papp e coll. 1996) e il ketoconazolo (Sluzewska e Nowakowska, 1994).
Anche lo shock elettroconvulsivo è efficace dopo una singola settimana di trattamento (Moreau e coll. 1995).
La mepiramina, un antistaminico, e l'atropina, un anticolinergico, sembrerebbero essere dei falsi positivi (Papp e coll. 1996), mentre l'ipsapirone, un agonista parziale 5-HT1a, diversamente dal buspirone, è inattivo nel modello CMS e ciò fa presupporre un caso di falso negativo.
Il trattamento con agonisti del recettore D2 dopaminergico è efficace nel contrastare l'anedonia indotta da CMS, mentre il trattamento con antagonisti D2 agisce in modo opposto (Muscat e coll. 1992; Papp e coll. 1993).
Alcuni studi hanno confermato che il modello CMS causa modificazioni, annullate dal trattamento con antidepressivi, della funzionalità di alcuni sistemi recettoriali. Per esempio, è stata dimostrata una diminuzione dei recettori D2/D3 nel nucleo accumbens e un aumento dei recettori 5-HT2 e ß-adrenergici nella corteccia cerebrale. Il CMS inoltre aumenta i recettori corticali 5-HT1A, ma questo effetto non è annullato dal trattamento cronico con antidepressivi (Papp e coll. 1994a, Papp e coll. 1994b; Willner e Papp, 1997).
Utilizzando tecniche atte a misurare in vivo il release di neurotrasmettitori, è stata dimostrata negli animali esposti al CMS, una diminuzione del rilascio di DA nel nucleo accumbens e nella corteccia prefrontale (Smadja, 1996).
Queste osservazioni nel complesso indicano che il modello CMS, oltre ad essere dotato di buon isomorfismo farmacologico, può fornire utili indicazioni sulla patogenesi della depressione.
Questo modello preclinico prevede la scelta di un fenotipo swim low-active (SwLo), cioè la scelta di animali che nel test di Porsolt presentano un tempo di immobilità superiore alla media. In base alla risposta comportamentale nel test del nuoto forzato, i ratti sono stati divisi in due gruppi a seconda della innata tendenza all'immobilità e, successivamente, incrociando più volte i ratti selezionati in funzione delle caratteristiche fenotipiche, si sono ottenuti due ceppi selezionati di ratti, una con elevato (swim low-active o SwLo) e l'altro con basso tempo di immobilità (swim high-active o SwHi).
L' osservazione comportamentale dei ratti SwLo e SwHi ha dimostrato che i ratti SwLo mostrano un comportamento passivo (Weiss, 1998; West e coll. 1999). Essi presentano una diminuzione della funzionalità dopaminergica e l'infusione di amfetamina nel cervello dei ratti SwLo causa soltanto un lieve aumento dell'attività motoria (West e coll. 1999).
Inoltre, la somministrazione di imipramina, venlafaxina e desipramina, fenelzina e bupropione, ma non di fluoxetina, sertralina e amitriptilina, provoca un aumento del comportamento attivo nei ratti SwLo sottoposti al test di nuoto forzato.
In accordo con questa risultati, si ritiene che il modello SwLo rappresenti un modello preclinico di depressione atipica (West e Weiss, 1998).
Gli antipsicotici
tipici (APT) sono stati utilizzati con successo per oltre 40 anni nel
trattamento di pazienti bipolari psicotici o in stato di agitazione
psicomotoria. Tali composti sono risultati essenziali nella risoluzione dei
casi più gravi, ad elevato rischio di mortalità, e il loro impiego ha reso
possibile la dimissione e la conseguente riabilitazione sociale dei pazienti
"psicotici cronici" istituzionalizzati. Bisogna tuttavia ricordare che, sebbene
questi farmaci posseggano una notevole efficacia e rapidità d'azione nel
controllo della sintomatologia psicotica e degli stati più gravi di agitazione
psicomotoria, non sembrano avere un'efficacia paragonabile a quella degli
stabilizzanti dell'umore nella profilassi delle ricorrenze del BD, in
particolare di quelle depressive. Tuttavia, nonostante l'utilità degli APT
nella terapia di mantenimento del disordine bipolare non sia mai stata
dimostrata, né indagata sistematicamente, il loro impiego è estremamente
diffuso, sia in monoterapia, che in combinazione con antidepressivi e
stabilizzanti dell'umore. Ciò malgrado sia ampiamente documentato come le
terapie combinate possano aumentare la frequenza e la gravità delle reazioni
avverse, a causa di un sinergismo reciproco nell'induzione di effetti
collaterali. Le linee guida proposte dalle varie organizzazioni internazionali
(APA, WHO, ecc.) suggeriscono all'unanimità che gli APT dovrebbero essere
utilizzati solo durante le fasi acute di malattia, sia per il rischio di
reazioni avverse, che per l'impatto negativo che il loro impiego protratto
potrebbe avere sul decorso a lungo termine del BD. Tuttavia, contrariamente a
quanto raccomandano gli esperti, molti pazienti bipolari assumono APT, sia in
maniera intermittente che continuativa, per lunghi periodi di tempo. Alcuni
studi hanno evidenziato (Sernyak e coll. 1997; Zarate e coll.
La definizione di antipsicotico "atipico" è stata sviluppata analizzando le caratteristiche della clozapina. L'elemento centrale dell' "atipicità", dal punto di vista clinico, è la proprietà di provocare effetti extrapiramidali in misura minore rispetto ai composti tipici; inoltre, fra le altre caratteristiche che gli APA dovrebbero possedere, bisogna ricordare la non induzione di iperprolattinemia, l'efficacia sui sintomi negativi della schizofrenia, così come sulle forme psicotiche resistenti agli APT. Fra tutti i farmaci definiti "atipici" solamente la clozapina sembra possedere tutte queste caratteristiche, mentre sia l'olanzapina che la quetiapina, pur avvicinandosi molto a questo profilo, ne presenterebbero solamente alcune. Il risperidone non sembra possedere molte delle caratteristiche di atipicità e per diversi aspetti non è dissimile dai farmaci definiti tipici. Anche per questi ultimi, infatti, la comparsa di effetti extrapiramidali è dose dipendente.
L'aloperidolo, prototipo di neurolettico "tipico", è dotato di un'elevata affinità per i recettori dopaminergici D2 mentre la clozapina, capostipite dei cosiddetti "atipici", mostra una bassa affinità per il medesimo sottotipo recettoriale. È opinione ampiamente condivisa che risposta clinica e comparsa di effetti collaterali risultino direttamente correlati alla percentuale di occupazione recettoriale D2 (Farde e coll. 1992). A tal proposito, Kapur e coll. (2000) hanno dimostrato come una saturazione recettoriale D2, pari ad almeno il 65% del totale, sia in grado di produrre una risposta clinica favorevole, così come non comparirebbero effetti collaterali di tipo extrapiramidale se la percentuale di saturazione si mantenesse al di sotto del 78%; inoltre, percentuali di occupazione recettoriale inferiori al 72% causerebbero soltanto incrementi minimi dei livelli di prolattina (Daskalakis e coll. 1998). Tali osservazioni sarebbero valevoli per tutti gli antipsicotici, tipici ed atipici; le differenze fra APT ed APA così come quelle, reciproche, fra le molecole appartenenti a quest'ultima classe farmacologica, sono state correlate alla diversa cinetica del legame recettoriale, come ad esempio alla durata di occupazione del recettore (Kapur e coll. 2000; Kapur e coll. 1998a; Jones e coll. 2000). Per quanto riguarda l'azione sui recettori serotoninergici 5-HT2A, molti "atipici" mostrano una percentuale di occupazione elevata, maggiore di quella per i D2, ma non correlata all'efficacia antipsicotica (Kapur e coll. 1998a; Kapur e coll. 1999; Nyberg e coll. 1997). Questa azione, tuttavia, non sembra rappresentare una condizione necessaria per l' "atipicità", anche in considerazione del fatto che neurolettici incontestabilmente "tipici", come la clorpromazina, possiedono anch'essi tale caratteristica (Kapur e coll. 1997; Martinot e coll. 1998). Inizialmente era stata formulata anche l'ipotesi secondo la quale le caratteristiche "atipiche" di un neurolettico potevano essere ricondotte al ruolo svolto dai recettori di tipo D4; in realtà, poiché l'affinità mostrata per tale sottotipo recettoriale da parte di APT come aloperidolo o clorpromazina, è più elevata rispetto a quella di "atipici" come clozapina od olanzapina (Seeman e coll. 1994), ed in ragione del fatto che farmaci selettivi per i D4 si sono mostrati privi di evidente azione antipsicotica (Bristow e coll. 1997), è da escludere che l'atipicità di un composto sia basata esclusivamente sull'affinità per i recettori D4, essendo tuttavia possibile un coinvolgimento reciproco dei recettori D2 e D4, ciascuno di essi dotato di un differente rapporto di affinità per i diversi principi attivi.
La clozapina (CLO) è stata scoperta in Svizzera nel 1959
dalla Sandoz-Wander. Per le sue caratteristiche stereochimiche tricicliche
peculiari, venne studiata inizialmente come antidepressivo, solo
successivamente si scoprì che la molecola era dotata di proprietà
antipsicotiche (Hippius, 1989). Nel
L' olanzapina (OLZ) è un antipsicotico atipico commercializzato dal 1996 per l'impiego nella schizofrenia. Negli ultimi anni, diversi studi controllati ne hanno confermato l'efficacia nel trattamento delle fasi acute maniacali o miste con o senza sintomi psicotici. Numerose indicazioni preliminari suggeriscono una possibile utilità di OLZ anche come stabilizzante dell'umore, per l'impiego nelle terapie di mantenimento e profilattiche del BD. Sia gli studi naturalistici, che i dati derivati dalle prove cliniche controllate su pazienti con disturbi dell'umore, pubblicati o presentati ai diversi congressi internazionali, sono numericamente maggiori per OLZ che per gli altri APA. Inizialmente, da alcune osservazioni cliniche è emerso come OLZ fosse dotata di proprietà antidepressive nei pazienti psicotici. Baker e coll. (1995) hanno riportato, infatti, che la somministrazione di 10 mg/die di OLZ era in grado di ridurre significativamente il punteggio totalizzato alla HAM-D di una vasta casistica di pazienti schizofrenici. Risultati analoghi sono stati riportati da Tollefson e coll. (1997) nella loro sperimentazione in doppio cieco, controllata verso placebo, condotta su di una popolazione di soggetti schizofrenici o schizoaffettivi. Successivamente in uno studio multicentrico parallelo di confronto in doppio cieco fra OLZ ed aloperidolo, della durata di 6 settimane (Thoen e coll. 1997), venne osservato che i pazienti schizoaffettivi, maniacali e misti, randomizzati ad OLZ, presentavano una riduzione maggiore dei "sintomi maniacali" alla BPRS rispetto a quelli randomizzati ad aloperidolo. Nello stesso studio, gli schizoaffettivi depressi trattati con OLZ totalizzavano punteggi inferiori rispetto al baseline alla Montgomery Ashberg Depression Rating Scale (MADRAS), mentre nei pazienti che avevano ricevuto aloperidolo veniva invece documentato un incremento medio di 6.63 punti rispetto al basale. In seguito a queste osservazioni, sono stati effettuati alcuni studi in aperto su pazienti con disturbi dell'umore resistenti ai trattamenti convenzionali. McElroy e coll. (1998) si sono proposti di valutare la risposta all'OLZ (dosaggio medio 14.1, sd = 7.2) in 14 pazienti con diagnosi di BD tipo I (secondo i criteri del DSM-IV) dimostratisi in precedenza poco responsivi al trattamento con stabilizzanti dell'umore o APT; 8 pazienti (57%) su 14 sono risultati "migliorati" o "molto migliorati" alla CGI-BP. Sharma e Pistor (1999) hanno riportato i dati relativi al trattamento con OLZ di 9 pazienti ambulatoriali con diagnosi di BD tipo I Episodio Misto secondo i criteri del DSM-IV; in precedenza tali pazienti non avevano risposto in maniera adeguata a stabilizzanti dell'umore, utilizzati in monoterapia oppure in combinazione con APT. Gli autori hanno riportato un miglioramento clinicamente significativo nella totalità dei casi trattati. Bates e coll. (1999) si sono recentemente occupati dell'impiego di OLZ nel trattamento della depressione psicotica; a tale scopo hanno somministrato OLZ in 15 pazienti depressi psicotici; tale popolazione è stata poi messa a confronto retrospettivamente con una popolazione di 15 depressi psicotici trattati con altri neurolettici. Dalle analisi di confronto è emerso che 10 pazienti (67%) su 15 trattati con OLZ risultavano "migliorati" o "molto migliorati" a confronto dei 4 (27%) su 15 cui erano stati somministrati altri antipsicotici. Più recentemente l'efficacia antimaniacale di OLZ è stata dimostrata con chiarezza in osservazioni controllate. Berk e coll. (1999) hanno confrontato OLZ e sali di litio, in doppio cieco e per una durata complessiva di 4 settimane, nel trattamento di 30 pazienti che soddisfacevano i criteri del DSM-IV per la diagnosi di mania acuta. Non sono state documentate differenze significative fra i due gruppi a confronto alla BPRS ed alla Mania Scale (MS); l'OLZ è però risultata superiore rispetto al litio alla CGI-severity scale al termine della quarta settimana di sperimentazione (Litio = 2.83, OLZ = 2.29; p = 0.025). Tohen e coll. (1999) hanno riportato i dati relativi al confronto fra OLZ e placebo nel trattamento della mania acuta. A tale scopo è stato condotto uno studio in doppio cieco, randomizzato, controllato verso placebo e della durata di 3 settimane. Dall'analisi dei risultati è emerso che il gruppo trattato con OLZ presentava un miglioramento clinicamente significativo sulla base dei punteggi totalizzati alla Young Mania Rating Scale; inoltre, la percentuale di pazienti che avevano risposto, era maggiore per l'OLZ (48.6%) rispetto al placebo (24.2%). OLZ è, tra gli APA, quello che presenta minori interazioni e può essere utilizzato con minori problemi in associazione con stabilizzanti, benzodiazepine ed antidepressivi. Gonzales-Pinto e coll. (2001) hanno impiegato OLZ, in combinazione con stabilizzanti in 44 pazienti maniaci ospedalizzati che soddisfacevano i criteri di McElroy e coll. (1996) per la diagnosi di Mania Disforica. Una buona risposta clinica era presente in 40 pazienti, con un miglioramento significativo della componente depressiva, in assenza di effetti collaterali o reazioni avverse di rilievo. Al fine di valutare efficacia e sicurezza di OLZ nel trattamento della mania acuta di bambini ed adolescenti, Frazier e coll. (2001) hanno condotto uno studio in aperto della durata di 8 settimane su di un campione di 23 soggetti bipolari (maniaci, ipomaniaci o misti) di età compresa fra 5 e 14 anni; gli autori hanno somministrato OLZ in monoterapia ed a dosaggi compresi fra 2,5 e 20 mg/die. Nei 22 pazienti (96%), che avevano completato la sperimentazione, la terapia con OLZ era in grado di promuovere un miglioramento significativo della sintomatologia clinica in assenza di effetti collaterali extrapiramidali. Vieta e coll. (2001) si sono proposti di stimare l'efficacia di OLZ in pazienti con BD scarsamente responsivo all'impiego dei soli stabilizzanti il tono affettivo. A tale scopo sono stati selezionati 23 soggetti con diagnosi di BD (Tipo I e Tipo II) già in trattamento con sali di litio, valproato o carbamazepina cui veniva aggiunta, a dosaggi crescenti, OLZ. L'osservazione ha avuto una durata di 43 settimane e il dosaggio medio di OLZ utilizzato è stato di 8,1 mg/die. L'analisi dei risultati ha permesso di rilevare una sensibile riduzione dei punteggi alla CGI con miglioramento della sintomatologia, sia depressiva che maniacale. Fra le reazioni avverse riportate si è osservata sonnolenza ed incremento ponderale. Sanger e coll. (2001) hanno portato a termine un'estensione a 43 settimane, in aperto, di una precedente loro sperimentazione in doppio cieco, controllata vs placebo e della durata di 3 settimane, relativa all'impiego di OLZ in 139 pazienti con BD Tipo I in fase espansiva. Dall'analisi dei risultati effettuata al termine del periodo di follow-up, OLZ si è dimostrata efficace, sia in monoterapia che in associazione a sali di litio e/o fluoxetina, nel migliorare la sintomatologia affettiva, ciò con associato un buon profilo di sicurezza. Il problema relativo all'eventuale presenza di predittori clinici di risposta all'OLZ in pazienti con disturbi dell'umore è stato affrontato da Zarate e coll. (1998) che hanno analizzato risultati relativi all'impiego di tale antipsicotico su di un campione di 150 pazienti. Gli autori hanno concluso che una risposta da "moderata" a "marcata" si correla più spesso con variabili quali la giovane età, una diagnosi di BD, una durata di malattia più breve, un periodo di ricovero più corto prima della somministrazione di OLZ, ed una durata della sperimentazione più lunga.
Fra i nuovi APA, il risperidone (RP) è stato il primo ad essere introdotto nella pratica clinica. Il farmaco appare dotato di tossicità inferiore a quella della CLO e, in diversi studi clinici comparativi su pazienti psicotici, è risultato parimenti efficace (Heinrich e coll. 1994). A dosi inferiori a 6 mg/die il farmaco possiede alcune delle caratteristiche di atipicità, tuttavia a dosi più elevate produce effetti extrapiramidali e fenomeni discinetici, al pari di altri APT. Anche per il RP è stata suggerita un'efficacia antimaniacale. Alcuni dati, tuttavia, indicano come il farmaco possa indurre un'esacerbazione dei sintomi maniacali, soprattutto quando somministrato in dosi elevate e senza uno stabilizzante dell'umore (Dwight e coll. 1994; Diaz, 1996). Il primo report disponibile in letteratura e relativo all'efficacia antimaniacale di RP è quello di Roose e coll. (1988). In tale esperienza, 3 pazienti psicotici in fase acuta su 4 trattati, con diagnosi di mania bipolare, mista e disturbo schizoaffettivo, avevano risposto ad una monoterapia a dosaggi medi di 4.9 mg/die. La durata media del follow up era di 8 mesi e mezzo. Successivamente sono state pubblicate altre osservazioni sull'efficacia di RP nel trattamento della mania acuta, in monoterapia o in combinazione. Singh e Catalan (1988) hanno segnalato una riduzione del punteggio alla YMRS pari al 77% nel corso di una monoterapia con RP, a dosi di 2-4 mg/die per una durata di 7-10 giorni, in 4 pazienti HIV positivi con sintomi acuti di mania psicotica. Goodnick (1995) ha riportato un miglioramento della sintomatologia maniacale in 2 pazienti bipolari trattati con RP in monoterapia o in associazione a sali di litio, a dosaggio medio di 6.5 mg/die. Infine, Thoen e coll. (1996) hanno riportato i dati relativi a 13 pazienti maniaci trattati in aperto, dei quali 10 (77%) presentavano una riduzione pari al 50% del punteggio alla YMRS e alla BPRS al termine della seconda settimana ed 8 del 75% al termine della sesta settimana. Altri studi in aperto, effettuati su pazienti ambulatoriali adulti oppure anziani con diagnosi di BD o Schizoaffettivo, hanno riportato percentuali di risposta al RP del 50% o anche superiori (Ghaemi e coll. 1997; Shaffer e Shaffer, 1996). Le osservazioni cliniche citate si riferiscono a casi clinici singoli o a piccole serie di pazienti bipolari trattati in aperto, spesso con combinazioni farmacologiche. Si comprende facilmente come i risultati ottenuti, per quanto incoraggianti, non consentono di trarre indicazioni sufficienti. Questo in considerazione del fatto che esistono anche osservazioni su casistiche simili che hanno fornito risultati in aperto contrasto con quelli sopra riportati. Sajatovic e coll. (1996) hanno, infatti, osservato che 5 (100%) pazienti in fase maniacale su 5 trattati con RP, avevano interrotto il farmaco per assenza di risposta o a causa degli effetti collaterali; in alcuni casi vi era stata una esacerbazione della sintomatologia espansiva. A tale proposito è necessario aggiungere che in letteratura sono riportati 5 "case reports" relativi a fasi maniacali indotte da RP (O'Cronin e Holt, 1995; Diaz, 1996; Tomlison, 1996; Barkin e Pais, 1997); inoltre, è stata descritta l'esacerbazione della sintomatologia maniacale in una serie di 6 soggetti schizoaffettivi trattati con RP (Diaz, 1996). Per quanto riguarda la depressione psicotica ed il disturbo schizoaffettivo-tipo depressivo, Hillert e coll. (1992) hanno riportato percentuali di risposta al RP pari al 70%, con un dosaggio medio di 6 mg/die; risultati positivi sono stati segnalati anche da Dwight e coll. (1994), che hanno somministrato RP ad 8 pazienti depressi schizoaffettivi oppure misti. Più recentemente, Vieta e coll. (2001) hanno riportato i dati relativi ad un trial clinico della durata di 6 settimane che prevedeva la somministrazione di RP in un campione di 102 pazienti affetti da disturbo schizoaffettivo, tipo bipolare. Avevano completato le 6 settimane di osservazione 95 pazienti ed è stato possibile concludere che RP possiede buone proprietà, sia antipsicotiche che stabilizzanti dell'umore; il farmaco è risultato inoltre ben tollerato, anche in relazione all'eventuale comparsa di sintomatologia extrapiramidale. Sempre di recente, Janicak e coll. (2001) si sono proposti di mettere a confronto l'efficacia e la sicurezza di RP rispetto all' aloperidolo in una sperimentazione randomizzata, in doppio cieco condotta su 62 pazienti schizoaffettivi, 29 tipo depressivo e 33 tipo bipolare. Al termine di tale ricerca è stato possibile rilevare che: a) l'aloperidolo produce effetti extrapiramidali maggiori rispetto al RP con conseguente percentuale maggiore di drop-out; b) non sono emerse differenze statisticamente significative fra i due gruppi in relazione all' efficacia sulla sintomatologia maniacale e psicotica; c) il RP è più efficace dell'aloperidolo sulla sintomatologia depressiva associata. In buona sostanza, il complesso di dati a disposizione sarebbe indicativo di una possibile efficacia del RP nelle forme maniacali o miste, da valutare meglio in studi controllati. A differenza degli APT il farmaco potrebbe avere proprietà antidepressive significative. La maggior parte dei dati si riferisce comunque ad osservazioni a breve termine. Nei trattamenti prolungati la tollerabilità di RP non è stata adeguatamente valutata nei pazienti con disturbi dell'umore. È possibile, infatti, che nel tempo RP tenda a produrre, analogamente ad alcuni APT, una maggiore incidenza di effetti extrapiramidali e di sintomi depressivi, con appiattimento e ritiro sociale. Keck e coll. (1995), mediante una revisione delle cartelle cliniche di 144 pazienti trattati consecutivamente con RP per un periodo di almeno due settimane, hanno riscontrato un miglioramento da "moderato" a "marcato" nei soggetti più giovani, con diagnosi di BD o schizoaffettivo-tipo depressivo, con una storia di malattia mentale più breve e con un minor numero di ospedalizzazioni nell'epoca antecedente l'inizio della terapia. Risultati sostanzialmente sovrapponibili erano stati riportati precedentemente da Dwight e coll. (Diaz, 1996).
La quetiapina (QTP) è un nuovo antipsicotico atipico da poco
in commercio sul mercato italiano. La letteratura psichiatrica internazionale
relativa all'uso di tale prodotto nel trattamento dei disturbi dell'umore è
ancora molto scarsa. Ghaemi e coll. (1999) hanno effettuato una revisione delle
cartelle cliniche di 6 pazienti con diagnosi di Disturbo Bipolare I secondo i
criteri del DSM-IV, che si erano dimostrati resistenti oppure intolleranti nei
confronti degli stabilizzanti tradizionali e ai quali era stata somministrata
QTP. Dall'analisi dei risultati è emerso che 2 pazienti su 6 avevano risposto
favorevolmente; il farmaco era ben tollerato e l'effetto collaterale
riscontrato più frequentemente era la sedazione. Zarate
e coll. (2000b) si sono proposti di valutare la possibile efficacia di QTP
nel trattamento dei disturbi affettivi psicotici e non, prefiggendosi anche di
identificare eventuali predittori di risposta. Sono stati trattati con QTP 145
pazienti con diagnosi di BD (maniacale, misto, depressivo), DDM con sintomi
psicotici, schizofrenia, disturbo schizoaffettivo (tipo bipolare o depressivo),
disturbo delirante o psicosi NAS; tutte le diagnosi sono state effettuate in
accordo con i criteri del DSM-IV. Dall'analisi dei risultati è emerso che i
soggetti bipolari (sia maniacali che misti o depressivi) e quelli
schizoaffettivi, tipo bipolare, presentavano tassi di risposta alla QTP
maggiori (> 74%) rispetto alla popolazione di schizofrenici, tuttavia
tali differenze non risultavano significative dal punto di vista statistico. Madhusoodanan
e coll. (2000) hanno somministrato QTP a 7 pazienti anziani ospedalizzati
(età compresa fra 61 e 72 anni) con sintomatologia riconducibile a
schizofrenia, disturbo schizoaffettivo o BD; a tutti erano stati somministrati
precedentemente antipsicotici convenzionali o altri APA. Il criterio di
risposta si è basato sulla semplice osservazione clinica delle eventuali
modificazioni comportamentali; 4 pazienti su 7 sono stati considerati
"responders". Fra gli effetti collaterali sono stati segnalati ipotensione
transitoria, irritabilità e sonnolenza; in due pazienti si è registrata
un'attenuazione di sintomi extrapiramidali preesistenti. Sajatovic e coll. (2001)
hanno condotto una sperimentazione in aperto di 12 settimane con lo scopo di
valutare l'efficacia e la sicurezza di QTP (range: 50-400 mg/die) in 20
soggetti con diagnosi di BD (n = 10) e disturbo schizoaffettivo
(n = 10), scarsamenre responsivi al trattamento con il solo
stabilizzante l'umore. Dall'analisi dei risultati è emersa la sostanziale
efficacia della strategia di potenziamento con QTP, come registrato dalle
diverse scale di valutazione impiegate (BPRS, HAM-D, YMRS). Il farmaco è
risultato, inoltre, ben tollerato. In
conclusione, da questa breve rassegna emerge che
Tabella 3. Profilo farmacodinamico degli antipsicotici atipici.
|
Farmaco |
Profilo farmacodinamico: blocco recettoriale |
|
Clozapina |
D1, D2, D4, 5-HT2, 1, H1, M1 |
|
Olanzapina |
D1, D2, 5-HT2, 1, H1, M1 |
|
Quetiapina |
D1, D2, 5-HT2, 1, H1, M1 |
|
Risperidone |
D2, 5-HT2, 1, 2, H1 |
Tabella 4. Affinità degli Antipsicotici Atipici per i recettori. D2 e 5-HT2A.
|
FARMACO |
D2 |
5-HT2A |
Rapporto 5-HT2A/D2 |
|
Clozapina | |||
|
Risperidone | |||
|
Olanzapina | |||
|
Quetiapina |
Tabella 5. Effetti collaterali principali degli antipsicotici atipici vs aloperidolo.
|
Aumento ponderale |
Anticolinergici |
PRL |
Agranulocitosi |
Ipotensione |
|
|
Clozapina | |||||
|
Olanzapina | |||||
|
Quetiapina | |||||
|
Risperidone | |||||
|
Aloperidolo |
Tabella 6. Impiego clinico degli antipsicotici atipici nelle diverse fasi dei disturbi dell' umore.
|
Clozapina |
Olanzapina |
Risperidone |
Quetiapina |
|
|
Ipomania/mania | ||||
|
Stati misti | ||||
|
Depressione bipolare | ||||
|
Rapida ciclicità | ||||
|
Profilassi |
Il disturbo bipolare (BD) è un disordine affettivo nel quale il paziente sperimenta non solo lo stato depressivo, caratterizzato da sintomi cognitivi, diminuzione del tono dell'umore, anedonia, anergia e perdita di motivazione, ma anche lo stato maniacale o ipomaniacale, in cui si manifesta una sintomatologia contropolare, con evidente aumento del tono dell'umore e agitazione psicomotoria. Negli individui affetti da BD gli episodi di depressione e di mania sono ricorrenti e si alternano tra loro con modalità e durata variabili. I farmaci stabilizzanti dell'umore rappresentano il trattamento di prima scelta dell'episodio maniacale acuto, nella depressione bipolare (talvolta in associazione con un antidepressivo) e nella terapia a lungo termine del disturbo bipolare per prevenire le ricadute. Attualmente i farmaci regolatori dell'umore sono il litio carbonato e farmaci anticomiziali come acido valproico, carbamazepina, lamotrigina, gabapentin e topiramato. Tuttavia, il ruolo che alcuni antipsicotici atipici come OLZ e QTP possono svolgere nel trattamento primario o aggiuntivo nelle diverse fasi del BD rappresenta un settore di ricerca emergente. Anche se nella letteratura clinica è presente una considerevole messe di dati a favore dell'impiego di questi farmaci nella mania acuta, ben poche sono le informazioni riguardanti la loro efficacia nel trattamento della depressione bipolare e nella prevenzione delle ricadute. Pertanto, un obiettivo della presente tesi è quello di confermare, se possibile, i risultati degli studi clinici che hanno segnalato l'attività antidepressiva degli antipsicotici atipici, valutando l'efficacia della QTP nel contrastare l'insorgenza dell'anedonia, un sintomo cardine della depressione umana, in un modello preclinico di depressione, lo stress cronico variato. Un altro importante obiettivo del lavoro sperimentale oggetto della tesi è quello di individuare i meccanismi molecolari responsabili di tale effetto mediante lo studio delle modificazioni della regolazione genica indotte dalla somministrazione del protocollo di stress cronico e dal trattamento con QTP nella corteccia frontale, un'area cerebrale di centrale importanza nell' eziopatogenesi dei disturbi affettivi. I risultati ottenuti potrebbero rivestire interesse in funzione del potenziale uso clinico della QTP come farmaco stabilizzante dell'umore.
Per la realizzazione di questa ricerca
sono stati impiegati ratti albini maschi del ceppo Wistar (Charles River,
Italia). Il peso medio dei ratti all'inizio della sperimentazione era di
Al termine del periodo di pre-stabulazione
i ratti sono stati alloggiati in gabbie trasparenti di plexiglas con superficie
minima del fondo di 935 cm² rivestita con una lettiera di tutolo di mais
depolverato. Sono stati predisposti tre animali per gabbia e la stabulazione è
stata condotta in condizioni standard con controllo costante di temperatura,
umidità e illuminazione. All'interno dello stabulario la temperatura è stata
mantenuta a 24 ±
Per tutta la durata della stabulazione i ratti hanno avuto libero accesso ad acqua e cibo e sono stati manipolati quotidianamente per addestrarli alla presa dell'operatore.
Tutte le pratiche di stabulazione, manipolazione e sperimentazione sono state condotte in conformità con la normativa CEE 86/609, OJL 358, 1 Dicembre 12, 1987; NH Guide for Care and Use of Laboratory Animals, NH Publications n° 85 - 23, 1985; D.L. 116/92.
In condizioni normali il ratto tende a prediligere il sapore di una soluzione zuccherina rispetto all'acqua.
La valutazione della preferenza al saccarosio ha richiesto la messa a punto di un protocollo preliminare, che consente di individuare ed eventualmente scartare gli animali che non mostrano tale preferenza.
Il protocollo prevede che gli animali
vengano prelevati dal loro abituale locale di stabulazione e alloggiati
singolarmente in gabbie di plexiglas nel luogo di sperimentazione. Ogni animale
viene fatto ambientare alla nuova situazione per 30 minuti. Al termine vengono
disposti sulla reticella di copertura di ogni gabbia due biberon della capacità di 250 ml
(Tecniplast-Gazzada S.a.r.l. - Varese), dotati di un cappuccio dosatore in
acciaio con un foro d'uscita del diametro di
I biberon vengono posizionati sempre all'estremità sinistra della grata di copertura, avendo cura di porre più esternamente quello contenente l'acqua che, nelle normali condizioni di stabulazione, è invece posizionato all'estremità destra.
Il peso dei due biberon viene rilevato prima di iniziare il test e successivamente ogni 2 ore per tutta durata della sperimentazione (26 ore); in questo modo è possibile monitorare il consumo individuale di acqua e di soluzione zuccherina. Al termine delle prime sei ore gli animali vengono mantenuti nelle stesse condizioni, ricevono il cibo e passano la notte nel locale di sperimentazione.
Le ultime due rilevazioni sul consumo di acqua e soluzione zuccherina vengono effettuate alla 24a ora e alla 26a ora dall'inizio del test.
Effettuata l'ultima rilevazione, la sperimentazione viene considerata conclusa; gli animali vengono riposti nelle loro gabbie abituali (è importante che siano ripristinati i gruppi di partenza per evitare inutili stress) e riportati nel luogo di stabulazione.
Per poter indurre l'anedonia negli animali, i ratti sono stati sottoposti ad un ciclo di stimoli di natura stressogena secondo il metodo proposto da Katz e coll. (1981) e opportunamente modificato da Ghi e coll. (1995).
Tale protocollo prevede diversi stimoli, più o meno intensi, che vengono proposti quotidianamente per un periodo di 42 giorni.
Gli stimoli che fanno parte del protocollo CMS sono:
Gli animali appartenenti ai gruppi CMS (sottoposti a stress cronico, trattati con salina) e TRATT (sottoposti a stress cronico, trattati con QTP), per tutta la durata della sperimentazione sono stati sottoposti a test settimanali di preferenza al saccarosio mediante i quali è possibile valutare l'insorgenza dello stato anedonico.
I test, della durata di 2 ore, sono stati effettuati secondo la procedura descritta in precedenza.
La preferenza al saccarosio è stata valutata anche negli animali del gruppo BAS, trattati con salina ma non sottoposti a protocollo di stress cronico.
Al termine del trattamento gli animali appartenenti ad ogni gruppo sperimentale sono stati sacrificati per il prelievo della corteccia frontale. Per effettuare il DNA microarray, il tessuto cerebrale proveniente da tre differenti animali è stato utilizzato per comporre un singolo campione. Tutti i campioni sono stati rapidamente immagazzinati alla temperatura di -80°C in attesa di successive analisi.
Per l'estrazione dell'RNA contenuto nel tessuto cerebrale dei ratti si utilizza una soluzione monobasica di guanidina tiocianato e fenolo, il TRI-REAGENT (SIGMA®), che è in grado di lisare i campioni di tessuto separando DNA, RNA e proteine. Si utilizza 1 ml di TRI-REAGENT/100 mg di tessuto.
Il tessuto viene pesato e posto all'interno di un omogenizzatore Potter, a cui si aggiunge l'esatta quantità di TRI-REAGENT; quindi si procede a omogeneizzare il tessuto, servendosi di un pestello, in modo da ottenere il lisato.
Si trasferisce l'omogenato così ottenuto in una provetta da 2 ml, si agita per alcuni secondi e il tutto viene lasciato a riposare per 5 minuti a temperatura ambiente.
Quindi si aggiunge cloroformio (0.2 ml cloroformio/ml di TRI-REAGENT usato), si agita vigorosamente in modo da ottenere una soluzione rosa pallido e si lascia a temperatura ambiente per 10-15 minuti.
La provetta contenente il tessuto
lisato e i due reagenti viene poi centrifugata a 12000 rpm per 15 minuti a
Il sopranatante viene prelevato e trasferito in una nuova provetta, dove vengono aggiunti 0.5 ml di isopropanolo/ml di TRI-REAGENT usato. La provetta, dopo agitazione manuale, viene lasciata a temperatura ambiente per 5-10 minuti.
Quindi si procede ad una seconda
centrifugazione a 12000 rpm per 10 minuti a
Con una pipetta, ponendo attenzione a non toccare con il puntale o asportare il pellet, si preleva ed elimina il sopranatante. A questo punto si aggiunge nella provetta contenente il pellet di RNA 1 ml di etanolo al 75%/ml di TRI-REAGENT usato in precedenza. Quindi si agita vigorosamente la provetta in modo manuale.
Infine si procede ad una terza
centrifugazione, questa volta a 10000 rpm per 5 minuti a
All'RNA così estratto si aggiungono 30
µl di acqua contenente dietilpirocarbonato (acqua DEPC), che è in grado di inibire
La soluzione di RNA è poi messa a scaldare a 55°-60° per 10-15 minuti.
Operando nel modo descritto, è stato
ottenuto RNA in soluzione, che può essere conservato nel tempo a
Al termine dell'estrazione dell'RNA è opportuno valutarne la purezza.
Vengono preparati i campioni di RNA estratto in provette da 1.5 ml, opportunamente numerate in modo da consentire l' identificazione dei campioni. In ogni provetta vengono posti 2 µl di campione e 198 µl di Acqua milliQ. E' preparata anche una provetta contenente solo 200 µl di acqua milliQ che rappresenta il bianco. Utilizzando una spettrofotometro e ponendo ad uno ad uno i campioni in una cuvetta di quarzo, si procede alla lettura dell'assorbanza dell'RNA estratto a due lunghezze d'onda, λ=260 nm e λ=280 nm.
Per analizzare la variazioni della regolazione genica abbiamo utilizzato il microarray, in particolare la tecnica dell'ibridazione in situ, sviluppata da Affimetrix. Tale tecnica è il risultato dell'interazione di due tecnologie particolari, la fotolitografia e la sintesi diretta in fase solida di oligonucleotidi.
La sintesi delle sonde oligonucleotidiche avviene direttamente su una superficie di supporto solido. Il supporto, costituito da un wafer di silicio, viene funzionalizzato con piccole sequenze di oligonucleotidi (oligo-starter), che hanno la caratteristica di possedere il gruppo reattivo protetto da gruppi fotosensibili. Grazie ad una maschera fotolitografica, è possibile indirizzare la luce in specifiche posizioni dell'array e così liberare i siti necessari per la sintesi della sequenza. Una volta deprotetti selettivamente i siti reattivi, è sufficiente incubare la superficie solida con desossiribonucleotidi protetti per allungare la catena in fase di sintesi.
Ripetendo il ciclo di deprotezione, grazie all'applicazione di maschere fotolitografiche diverse, e di incubazione, è quindi possibile aggiungere nucleotidi diversi in posizioni diverse e sintetizzare tutte le sonde necessarie per l'analisi di un genoma di interesse.
I targets, ovvero gli acidi nucleici da ibridizzare alle catene di cDNA ancorate al supporto solido, sono normalmente ottenuti dalla marcatura con molecole fluorescenti del mRNA proveniente da un dato organismo. Le sonde e i targets vengono poi messi a contatto per promuovere la reazione di ibridazione in situ. Quindi l'array viene passato attraverso uno scanner per la misurazione dei segnali fluorescenti. L'intensità dei pixel di ciascuna immagine è proporzionale al numero di molecole di tracciante presenti sullo spot e, quindi, al numero di sonde che hanno ibridizzato le sonde ancorate al supporto. Di fatto, livelli diversi di fluorescenza indicano livelli diversi di ibridizzazione e quindi di espressione genica. Il segnale rilevato dallo scanner viene poi sottoposto ad algoritmi di filtrazione e di pulizia del segnale e convertito in valori numerici.
Nel complesso il microarray, in qualità di esperimento di analisi dei profili di espressione genica, fornisce come risultato una matrice di dati, in cui le righe rappresentano i geni monitorati e le colonne corrispondono alle diverse condizioni sperimentali (punti temporali, condizioni fisiologiche, tessuti). Ogni elemento della matrice rappresenta quindi il livello di espressione di un particolare gene in uno specifico stato fisiologico.
Per realizzare la tecnica Real-Time utilizziamo il Brillant SYBR Green QPCR Master MIX della Stratagene (Codice prodotto 600548).
Per ogni singola applicazione di Real-Time è necessario preventivamente settare i primers senso (SE) e antisenso (AS), sia per il gene di controllo (ACTINA o GADPH), sia per il GOI (Gene of Interest).
Al fine di evidenziare eventuali errori dipendenti dall'azione dell'operatore, è opportuno che il settaggio dei primers venga proposto in doppio.
Inoltre è indispensabile che tutte le operazioni sperimentali vengano effettuate rigorosamente in ghiaccio.
I primer vengono settati predisponendo soluzioni a concentrazioni variabili, 50 nM, 150 nM e 300 nM.
Secondo il protocollo della Stratagene il volume finale della miscela di reazione dovrà essere di 25 μl.
Le diverse soluzioni madre di primers (nelle quantità indicate nella tabella della pagina seguente) vanno aggiunti alle mix di reazione in singole provette (STARGENE 401428-tubi 401425-tappi), in modo tale da determinare tutte le combinazioni possibili di concentrazione.
|
CALCOLO MIX PRIMER PER REAL TIME da 3 μM |
||||||
|
Primer SE |
Primer AS | |||||
|
μM |
μM |
μM |
μM |
μM |
μM |
Acqua da aggiungere |
|
(12.125-(AS+SE)-(cDNA)) |
||||||
|
(12.125-(AS+SE)-(cDNA)) |
||||||
|
(12.125-(AS+SE)-(cDNA)) |
||||||
|
(12.125-(AS+SE)-(cDNA)) |
||||||
|
(12.125-(AS+SE)-(cDNA)) |
||||||
|
(12.125-(AS+SE)-(cDNA)) |
||||||
|
(12.125-(AS+SE)-(cDNA)) |
||||||
|
(12.125-(AS+SE)-(cDNA)) |
||||||
|
(12.125-(AS+SE)-(cDNA)) |
||||||
All'interno di ogni tubo di reazione pongo 0.25 μl di campione.
A questo punto è possibile preparare in una provetta sterile la mix di reazione che, per ogni singolo campione, è così formata:
12.5 μl di mastermix + 0.375 μl di Rox = 12.875 μl
Quindi si aggiunge la mix di reazione al campione contenuto nel tubo.
Per procedere nella realizzazione della Real-Time è necessario preparare una curva di taratura che consente di risalire alla concentrazione di DNA presente nel campione. Considerando di analizzare due geni, la curva va riproposta in doppio o in triplo, per tutti i geni in esame. E' importante utilizzare uno standard di riferimento come l'actina o il GADPH.
Per preparare una corretta curva di taratura devo considerare il range di diluizione in cui ricade il campione di cDNA sottoposto a real-time.
Il campione in analisi, nel protocollo di real-time utilizzato durante il nostro studio, è diluito di 50 volte. Quindi devo creare una retta di taratura che necessariamente ricada entro tale valore di diluizione.
Si preparano tre diverse strisce di tubi da RT, una per l'actina e due per i geni di interesse, come mostrato nella figura seguente.

Le strisce sono costituite da 8 tubi, due per ciascuna diluizione da effettuare.
Si preparano 4 tubi e, dopo averli numerati, si procede come di seguito indicato:
TUBO 1 = 18 μl di cDNA + 45 μl di Acqua MilliQ = 63 μl tot.
In questo modo si ottiene una diluizione di 3,5 volte.
A questo punto si prelevano 7.125 μl di campione dal tubo 1 e si distribuiscono in ognuno dei tubi di reazione (2 per actina e 2 per ogni GOI).
TUBO 2 = si prelevano 18 μl dal tubo 1 e si diluiscono ancora di 3.5 volte, aggiungendo sempre 45 μl di acqua (diluizione finale: 3.5
A questo punto si prelevano 7.125 μl di campione dal tubo 2 e si distribuiscono in ognuno dei tubi di reazione (2 per actina e 2 per ogni GOI).
TUBO 3 = si prelevano 18 μl dal tubo 2 e si diluiscono ancora di 3.5 volte (diluizione finale: 12.25
A questo punto si prelevano 7.125 μl di campione dal tubo 3 e si distribuiscono in ognuno dei tubi di reazione (2 per actina e 2 per ogni GOI).
TUBO 4 = si prelevano 18 μl dal tubo 3 e si diluiscono ancora di 3.5 volte (diluizione finale: 42.875
A questo punto prelevo 7.125 μl del campione del tubo 3 e si distribuisco in ognuno dei tubi di reazione (2 per actina e 2 per GOI).
Una volta conclusa l'operazione di distribuzione si prepara la mix di reazione, tenendo presente che la quantità di cDNA aggiunto è stata di 7.125 μl e che il volume finale in ogni tubo deve essere di 25 μl.
PREPARAZIONE DELLA MIX DI REAZIONE PER L'ACTINA:
MIX 2X = 12.5 (n° campioni + 1)
ROX = 0.375 (n° campioni + 1)
PRIMER SE = 2.5 (n° campioni + 1)
PRIMER AS = 2.5 (n° campioni + 1)
In ogni singolo tubo devono essere aggiunti 17.825 μl di mix di reazione per actina.
PREPARAZIONE DELLA MIX DI REAZIONE PER GOI:
MIX 2X = 12.5 (n° campioni + 1)
ROX = 0.375 (n° campioni + 1)
PRIMER SE = 2.5 (n° campioni + 1)
PRIMER AS = 2.5 (n° campioni + 1)
In ogni singolo tubo devono essere aggiunti 17.825 μl di mix di reazione per GOI.
Si preparano tre strisce di tubi, una per l'actina e una per ognuno dei due GOI.

In ogni singolo tubo vengono posti 0.25 μl di campione.
Nel tubo 7 (bianco) non va aggiunto il campione, ma solo l'acqua in quantità finale di 7.125 μl. Quest'ultimo tubo rappresenta il controllo, per verificare che i campioni non siano contaminati o che non si siano verificati errori nella preparazione.
A questo punto è possibile preparare la mix di reazione per l'actina e per GOI, che, per ogni campione, sarà composta come qui di seguito indicato.
ACTINA:
MIX 2X = 12.5 μl
ROX = 0.375 μl
PRIMER SE = 2.5 μl
PRIMER AS = 2.5 μl
ACQUA = 6.875 μl
GOI:
MIX 2X = 12.5 μl
ROX = 0.375 μl
PRIMER SE = 2.5 μl
PRIMER AS = 2.5 μl
ACQUA = 6.125 μl
A questo punto, dopo aver alloggiato i tubi di reazione nel termociclatore per Real-Time, parte il programma di amplificazione, al termine del quale è possibile verificare la quantità di cDNA presente in ogni tubo.
Kary Mullis nel 1993 ricevette il premio Nobel per aver messo a punto la tecnica della Reazione a Catena della Polimerasi (PCR).
Grazie all'azione dell'enzima retrotrascrittasi inversa, è possibile retrotrascrivere l'RNA a singolo filamento ed ottenere i corrispondenti cDNA a doppia elica. I cDNA sono poi utilizzati come stampo per l'amplificazione della sequenza di DNA e per la valutazione dell'espressione genica.
Utilizzando una soluzione contenente i
cDNA, gli oligonucleotidi, i primers (in grado di legare in modo specifico la
sequenza di DNA), i desossiribonucleotidi trifosfati, gli ioni magnesio e
I primers vengono creati sinteticamente sulla base della sequenza di DNA oggetto di studio al fine di permettere l'appaiamento e il legame dei primers stessi alle regioni complementari di cDNA.
L'RNA a singolo filamento, estratto dal tessuto cerebrale di ratto, deve essere retrotrascritto in modo da ottenere il doppio filamento di cDNA, che sarà poi utilizzato per la successiva reazione di amplificazione tramite PCR.
Durante lo studio ci siamo avvalsi dell'uso del KIT PROOMEGA® (ImProm-II Reverse Trascription System).
In tubi sterili e immersi nel ghiaccio si preparano i campioni di RNA e, per ogni campione, si aggiungono nel seguente ordine:
1) RNA totale 0.1-5 ng = 2 µl. E' necessario che l'RNA totale sia comunque superiore a 1 mg/reazione.
2) Oligo(dT)15 primer = 1 µl. Sono necessari 0.5 mg Oligo(dT)15 primer/r azione.
3) Nuclease Free Water = 2 µl. Il protocollo consente l'aggiunta di Nucleare Free Water fino ad un volume massimo di 5 ml, in funzione della quantità di RNA totale utilizzato.
Il volume finale di ogni campione di RNA da retrotrascrivere è di 5 ml.
I tubi di reazione vengono poi
riscaldati a
Nel frattempo si prepara la miscela di reazione, avendo cura di aggiungere solo in ultimo gli enzimi necessari per la retrotrascrizione, in quanto soggetti a facile degradazione. Per ogni campione si preparano 15 µl di miscela di reazione aggiungendo nell'ordine:
1) Nuclease Free Water = 7.3 µl. Il protocollo consente l'aggiunta di Nucleare Free Water fino ad un volume massimo di 15 ml.
2) Improm-II 5X Reaction Buffer = 4 µl
3) MgCl2,
4) dNTPs Mix 10mM = 1 µl. La
concentrazione finale di dNTPs deve essere di
5) l'enzima Recombinant RNasi Ribonuclease Inhibitor = 0.5 µl
6) l'enzima Improm-II Riverse Transcriptase = 1 µl
A questo punto, in ogni singola provetta contenente il campione di RNA precedentemente preparato (5 µl), si aggiungono 15 µl di miscela di reazione per ottenere un volume finale di 20 µl.
Le provette vengono poste nel termociclatore dove vengono sottoposte ai 3 seguenti passaggi:
1°:
2°:
3°:
Il passaggio finale ha lo scopo di inattivare gli enzimi.
Procedendo in questo modo, abbiamo trasformato il filamento singolo di RNA estratto in un doppio filamento di cDNA, pronto per essere amplificato tramite PCR.
Dopo retrotrascrizione dell'RNA, i
campioni di cDNA possono essere conservati nel tempo a
Per l'amplificazione del cDNA retrotrascritto abbiamo utilizzato la tecnica PCR (Reazione a Catena della Polimerasi).
E' necessario che tutte le operazioni di preparazione previste dal protocollo di amplificazione vengano eseguite rigorosamente in ghiaccio.
In una provetta da 1.5 ml si prepara la
miscela di reazione, avendo cura di aggiungere solo in ultimo
Buffer 10X (EPPENDORF®) = 2.5 µl
Mix Primer AS+SE 5 μM = 2.5 µl. La concentrazione finale di Mix Primer AS+SE deve essere di 0.5 µM.
MgCl2
dNTPs
Taq Polimerasi (EPPENDORF®) = 0.1 µl
Quindi si aggiunge alla miscela di reazione acqua milliQ in quantità tale da ottenere un volume finale di miscela di 20 µl per ogni campione (20-6.6 = 13.4 µl acqua milliQ).
A questo punto, in ogni singola provetta, contenente 5 µl di campione di cDNA precedentemente preparato, vengono aggiunti 20 µl di miscela di reazione per ottenere un volume finale di 25 µl.
Le provette contenenti cDNA e miscela di reazione vengono poste nel termociclatore dove saranno sottoposte a cicli termici.
CARATTERISTICHE PRIMERS
Bax SENSO - GAGACACCTGAGCTGACCTT -
|
Len |
MW |
Tm: 61.05° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Bax ANTISENSO - ATCTTTGTGGCTGGAGTCCT
|
Len |
MW |
Tm: 62.62° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Bad SENSO - TTGAGCCGAGTGAGCAGGAA -
|
Len |
MW |
Tm: 68.43° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Bad ANTISENSO - CTATGGCCGTGAGCTCCGAA - VEDERE
|
Len |
MW |
Tm: 69.52° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Bcl2 SENSO - CCGGGAGATCGTGATGAAGT
|
Len |
MW |
Tm: 66.28° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Bcl2 ANTISENSO -TGGTGGACAACATCGCTCTG
|
Len |
MW |
Tm: 67.44° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Bcl2l1 SENSO - CTGTGGAGATCCCTAACTGC -
|
Len |
MW |
Tm: 61.25° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Bcl2l1 ANTISENSO - CAGCCTGTGTGTTTACT -
|
Len |
MW |
Tm: 60.78° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Tieg SENSO - ACAGTTTGCT TCCAGGGACG -
|
Len |
MW |
Tm: 66.36° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Tieg ANTISENSO AGGTCTTCTCAGCCAGCTCC -
|
Len |
MW |
Tm: 64.94° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Ebag9 SENSO - AGCAGACAGACGTGGAGGA -
|
Len |
MW |
Tm 63.83° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Ebag9 ANTISENSO - CCCAGATGGTAGCACAGGT - VEDERE
|
Len |
MW |
Tm 62.61° C |
GC |
Sec. tr.: None |
Primer Dimer: No |
Sgk SENSO - TGCTCGAAGTACCCTCACCT -
|
Len |
MW |
Tm: 63.74° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Sgk ANTISENSO - TTAATGGCGGAGAGCTGTTC -
|
Len |
MW |
Tm: 64.08° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Stk17b SENSO - GGCCACAGACTTCATCCAGA - VEDERE
|
Len |
MW |
Tm: 65.31° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Stk17b ANTISENSO - CGATTCGATGACTCCTTGCC -
|
Len |
MW |
Tm: 66.85° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Nlgn3 SENSO - TCATGCTAGGCGTCAACCAG -
|
Len |
MW |
Tm: 66.25° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Nglgn3 ANTISENSO - ACCATCACTGCCAGAGCCTC -
|
Len |
MW |
Tm: 66.77° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Itgam SENSO - AGCTGGTGATCACGTGTTCC - VEDERE
|
Len |
MW |
Tm: 65.21° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Itgam ANTISENSO - TGCAGTCATCTCGAGGAACC - VEDERE
|
Len |
MW |
Tm: 65.44° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Itga6 SENSO - GGCCAGTTGTGCTTGCTCTA -
|
Len |
MW |
Tm: 65.36° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Itga6 ANTISENSO - TTCTGTGATGGCCGATTGAG -
|
Len |
MW |
Tm: 66.14° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Hes5 SENSO - ACCGCATCAACAGCAGCATT - VEDERE
|
Len |
MW |
Tm: 67.88° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Hes5 ANYISENSO - GCACCAGGACTACAGCGAGG - VEDER
|
Len |
MW |
Tm: 66.58° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Cldn10 SENSO - ATCATCGCCTTCGTCGTCTC -
|
Len |
MW |
Tm: 66.55° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Cldn10 ANTISENSO - GCATCATTGGCGGTGTCATA -
|
Len |
MW |
Tm: 66.76° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Cdh1 SENSO - TCGCCTACACCATCCTCAGC -
|
Len |
MW |
Tm: 67.54° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Cdh1 ANTISENSO - TTGAGAATGAGGTCGGTGCC -
|
Len |
MW |
Tm: 67.42° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Cdh7 SENSO - TTGTGGAAGACGTGGATGAG -
|
Len |
MW |
Tm: 63.79° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Cdh7 ANTISENSO - GCTCAGCCAGGACAGGTTAT -
|
Len |
MW |
Tm: 63.13° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Cdh22 SENSO - GAGTACACGGGGACAGAACC -
|
Len |
MW |
Tm: 63.36° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
Cdh ANTISENSO - AAGAAAGTGGCAGTGGAGGA -
|
Len |
MW |
Tm: 63.73° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
ACTINA SENSO - ACATCGGCAAAGACTTGTACG
|
Len |
MW |
Tm: 61.10° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
ACTINA ANTISENSO - AGCATTTGCGGTGGACGATGG
|
Len |
MW |
Tm: 73.2° C |
GC |
Sec. Str.: None |
Primer Dimer: No |
FASI DEL CICLO PCR
Fase di Denaturazione: la soluzione contenente i cDNA
da replicare, i desossiribonucleotidi trifosfati, gli ioni magnesio, il primer
e
Fase di Annealing: la temperatura viene abbassata fino alla temperatura di annealing al fine di permettere il legame dei primer alle regioni complementari dei filamenti di cDNA denaturati.
Fase di Estensione: la temperatura viene aumentata
fino a 65-
Il singolo ciclo appena descritto viene ripetuto generalmente per circa 20-30 volte.
I cicli di lavoro per i geni in esame sono i seguenti:

Al termine dei cicli di amplificazione previsti per i geni in esame, i campioni di DNA possono essere conservati nel tempo a -20°C.
Attraverso la corsa elettroforetica è possibile valutare le differenze quantitative dell'espressione dei geni di interesse a partire dal DNA amplificato proveniente dai diversi campioni di ratto (basale, stressato o CMS e trattato con quetiapina).
Per la corsa elettroforetica dei campioni è stato utilizzato un gel di agarosio al 2% disciolto in una soluzione di TBE.
PREPARAZIONE TBE 100 ML 10X
In un becker contenente un'ancoretta sono stati aggiunti:
Si è portato a volume con 100 ml di acqua milliQ e si è sciolto il tutto utilizzando un agitatore.
La concentrazione di utilizzo del TBE è 0.5X.
PREPARAZIONE DEL GEL DI AGAROSIO 2%
In un becker sono stati aggiunti:
Il becker contenente il TBE e l'agarosio è portato a ebollizione in forno a microonde, in modo da sciogliere l'agarosio.
Infine si aggiungono 2 µl di etidio bromuro (SIGMA®) e si cola la miscela nell'apposito apparecchio per la corsa elettroforetica, dopo aver inserito opportunamente i pettini.
Una volta avvenuta la solidificazione del gel, si aggiunge il tampone di corsa nell'apparecchio elettroforetico e si tolgono i pettini che, intrappolati nel gel, hanno creato i pozzetti necessari per il caricamento dei campioni.
Per poter procedere al caricamento negli appositi pozzetti, i campioni e il marker devono essere preparati.
Il marker (θ174) consente di
identificare i campioni di DNA aventi dimensioni che vanno da
Invece ai campioni (ogni campione = 5 μl) deve essere aggiunto 1 μl di Loading Day.
Una volta che i campioni e il marker sono stati opportunamente caricati, si può dare inizio alla corsa elettroforetica a 90-100 V.
STRESS CRONICO VARIATO
Gli effetti provocati dalla somministrazione cronica di salina, QTP o AMI sul consumo di saccarosio di ratti naive o sottoposti al protocollo di stress cronico sono illustrati nella figura 2.
L'analisi della varianza (ANOVA) a disegno misto, applicata ai dati sperimentali ottenuti, indica un effetto significativo della variabile indipendente "trattamento" (F 3/44 = 96.4; p < 0.001) e della variabile dipendente "data del test di preferenza al saccarosio" (F 5/220 = 11.1; p < 0.001). Anche l'interazione tra le due variabili è statisticamente significativa (F 15/220 = 8.2;
p < 0.001). La successiva analisi
indica che il gruppo trattato con salina e sottoposto al protocollo CMS riduce
progressivamente il consumo di saccarosio rispetto al basale, stabilizzandosi
su valori pari a circa il 50%, intorno alla terza settimana dall'inizio della
sperimentazione (F 1/44 = 281; p < 0,001). Sia il trattamento con QTP 2
mg/kg/die (F 1/44 = 65; p < 0.001) che quello con AMI 2 mg/kg/die (F 1/44 =
112; p < 0.001) sono in grado di contrastare la progressiva riduzione del
consumo di saccarosio indotta dallo stress cronico. L'effetto della AMI compare
intorno alla quarta settimana, mentre quello della QTP compare circa una
settimana dopo. Pertanto, la differenza statisticamente significativa tra il
gruppo trattato con QTP e quello trattato con
 Figura
2. Effetto del trattamento cronico con QTP e AMI sull'anedonia indotta dal
protocollo di stress cronico variato
Figura
2. Effetto del trattamento cronico con QTP e AMI sull'anedonia indotta dal
protocollo di stress cronico variato
DNA MICROARRAY
Il numero totale di geni presi in considerazione è 31099.
Le modificazioni nella trascrizione dei singoli geni nella corteccia frontale, provocate dallo stress cronico e dal trattamento con QTP e calcolate mediante algoritmo GCRMA, sono riportate in figura 3.

Figura 3.
In seguito a somministrazione del protocollo CMS, il numero di geni che mostra una variazione di ± 1.5 volte rispetto alle condizioni basali (confronto gruppo BAS Vs gruppo CMS) è 325 (figura 4.)

Figura 4.
Tra questi, 126 sono up-regolati (fold change da +
La successiva analisi statistica, effettuata su questi geni mediante il T-Test di Welch (livello di significatività statistica p=0.01) ha indicato una differenza significativa in 63 geni, le cui caratteristiche sono riportate nelle tabelle 7. e 8. e nella figura 5.
Tabella 7. Geni up-regolati dallo stress cronico nella corteccia frontale del ratto
|
Genbank |
QTP |
Gene Symbol |
Gene title |
|
NM_138838 |
Pou3f1 |
POU domain, class 3, transcription factor 1 |
|
|
NM_012551 |
Egr1 |
early growth response 1 |
|
|
NM_053633 |
Egr2 |
early growth response 2 |
|
|
NM_024388 |
Nr4a1 |
nuclear receptor subfamily 4, group A, member 1 |
|
|
NM_017352/// NM_031628 |
Nr4a3 |
nuclear receptor subfamily 4, group A, member 3 |
|
|
NM_017232 |
Ptgs2 |
prostaglandin-endoperoxide synthase 2 |
|
|
NM_017313 |
RABIN3 |
RAB3A interacting protein |
|
|
NM_024125 |
Cebpb |
CCAAT/enhancer binding protein (C/EBP), beta |
|
|
XM_221496 |
Esrrbl1_predicted |
estrogen-related receptor beta like 1 (predicted) |
|
|
NM_001024784 |
Nfx1_predicted |
nuclear transcription factor, X-box binding 1 (predicted) |
|
|
NM_080887 |
Txnl1 |
thioredoxin-like (32kD) |
|
|
NM_019242 |
Ifrd1 |
interferon-related developmental regulator 1 |
|
|
NM_139216/// NM_139217 |
Kcnc2 |
potassium voltage gated channel, Shaw-related subfamily, member 2 |
|
|
NM_133317 |
Tob1 |
transducer of ERBB2, 1 |
|
|
NM_031135 |
Tieg |
TGFB inducible early growth response |
|
|
XM_214331 |
Klhl2_predicted |
kelch-like 2, Mayven (Drosophila) (predicted) |
|
|
XM_224894 |
Cnot7_predicted |
CCR4-NOT transcription complex, subunit 7 (predicted) |
|
|
XM_345674 |
Cfl2_predicted |
cofilin 2, muscle (predicted) |
|
|
XM_342731 |
Sumf1_predicted |
sulfatase modifying factor 1 (predicted) |
|
|
NM_001009665 |
Ebag9_predicted |
estrogen receptor-binding fragment-associated gene 9 (predicted) |
|
|
XM_232640 |
Sox17_predicted |
SRY-box containing gene 17 (predicted) |
|
|
NM_153821 |
Prrx1 |
paired related homeobox 1 |
|
|
NM_145085 |
Pcyox1 |
chloride ion pump-associated 55 kDa protein |
|
|
NM_001009661 |
Wbp11_predicted |
WW domain binding protein 11 (predicted) |
|
|
XM_342300 |
Zfp364_predicted |
zinc finger protein 364 (predicted) |
|
|
NM_133402 |
Nap1l3 |
nucleosome assembly protein 1-like 3 |
|
|
XM_219292 |
sp31_predicted |
ubiquitin specific protease 31 (predicted) |
|
|
NM_001013250 |
Ddx21a_predicted |
DEAD (Asp-Glu-Ala-Asp) box polypeptide 21a (predicted) |
|
|
NM_012957 |
Gabrb2 |
gamma-aminobutyric acid receptor, subunit beta 2 |
|
|
NM_145678 |
Vps4a |
vacuolar protein sorting 4a (yeast) |
|
|
NM_022197 |
Fos |
murine osteosarcoma viral oncogene homolog |
|
|
NM_021836 |
Junb |
Jun-B oncogene |
|
|
NM_138921 |
Eml2 |
echinoderm microtubule associated protein like 2 |
|
|
NM_013150 |
Nrcam |
neuron-glia-CAM-related cell adhesion molecole |
|
|
NM_031334 |
Cdh1 |
Cadherin 1 |
|
|
NM_017171 |
Prkce |
Protein Kinase c, epsilon |

Figura 5.
Tra questi, 244 sono up-regolati (fold change da +
La successiva analisi statistica, effettuata su questi geni mediante il T-Test di Welch (livello di significatività statistica p=0,01) ha indicato una differenza significativa in 98 geni (figura 6.)
Figura 6.
 Fra i geni up-regolati dallo stress cronico, 28 (Pou3f1, Egr1, Nr4a1, Nr4a3,
Ptgs2, Esrrbl1_predicted, Nfx1_predicted, Txnl1, Ifrd1, Tob1, Tieg, Klhl2_predicted,
Cnot7_predicted, Cfl2_predicted, Sumf1_predicted, Ebag9_predicted, Pcyox1, Wbp11_predicted,
Zfp364_predicted, Nap1l3, sp31_predicted, Gabrb2, Vps4a, Fos, Junb, Nrcam,
Cdh1, Prkce) subiscono una regolazione opposta e ritornano presumibilmente alle
condizioni rilevate negli animali non anedonici, 2 geni (Egr2 e Prrx1)
subiscono una regolazione nello stesso verso e i rimanenti 6 non vengono
modificati (RABIN3, Cebpb, Kcnc2, Sox17_predicted, Ddx21a_predicted, Eml2) dal
trattamento con QTP.
Fra i geni up-regolati dallo stress cronico, 28 (Pou3f1, Egr1, Nr4a1, Nr4a3,
Ptgs2, Esrrbl1_predicted, Nfx1_predicted, Txnl1, Ifrd1, Tob1, Tieg, Klhl2_predicted,
Cnot7_predicted, Cfl2_predicted, Sumf1_predicted, Ebag9_predicted, Pcyox1, Wbp11_predicted,
Zfp364_predicted, Nap1l3, sp31_predicted, Gabrb2, Vps4a, Fos, Junb, Nrcam,
Cdh1, Prkce) subiscono una regolazione opposta e ritornano presumibilmente alle
condizioni rilevate negli animali non anedonici, 2 geni (Egr2 e Prrx1)
subiscono una regolazione nello stesso verso e i rimanenti 6 non vengono
modificati (RABIN3, Cebpb, Kcnc2, Sox17_predicted, Ddx21a_predicted, Eml2) dal
trattamento con QTP.
Al contrario, tra i geni down-regolati dallo stress cronico, 21 (LOC306340, Dlc1, Eif2b5, Dcc, Clic1, Gna12, Snrp70_predicted, Itga6, Cdh22, Fgfr1, U2af2_predicted, Nr2f6, Camk2a, Phactr1, Homer1, Grik5, Vegf, Atp2a2, Prkcb1, Ppp3ca, Bcl2l1) subiscono una regolazione opposta, 3 geni (Mbp, Csf1 e Grid1) subiscono una regolazione nello stesso verso e i rimanenti 3 non vengono modificati (Gpr135, Mpk10, Rab21) dal trattamento con QTP.
Tabella 8. Geni down-regolati dallo stress cronico nella corteccia frontale del ratto
|
Genbank |
QTP |
Gene Symbol |
Gene title |
|
XM_224715 |
LOC306340 |
similar to mKIAA1623 protein |
|
|
XM_341444 |
Dlc1 |
deleted in liver cancer 1 |
|
|
NM_138866 |
Eif2b5 |
eukaryotic translation initiation factor 2B, subunit 5 epsilon |
|
|
NM_012841 |
Dcc |
deleted in colorectal carcinoma |
|
|
NM_001033968/// NM_053609 |
Clic1 |
Chloride intracellular channel 1 |
|
|
NM_031034 |
Gna12 |
guanine nucleotide binding protein, alpha 12 |
|
|
XM_341857 |
Snrp70_predicted |
U1 small nuclear ribonucleoprotein polypeptide A (predicted) |
|
|
NM_001025289 /// NM_001025291 /// NM_001025292 /// NM_001025293 /// NM_001025294 /// NM_017026 |
Mbp |
myelin basic protein |
|
|
XM_215984 |
Itga6 |
integrin, alpha 6 |
|
|
NM_019161 |
Cdh22 |
cadherin 22 |
|
|
NM_181771 |
Gpr135 |
G protein-coupled receptor 135 |
|
|
NM_024146 |
Fgfr1 |
Fibroblast growth factor receptor 1 |
|
|
NM_023981 |
Csf1 |
Colony stimulating factor 1 |
|
|
XM_218195 |
U2af2_predicted |
U2 small nuclear ibonucleoprotein auxiliary factor (U2AF) 2 (predicted) |
|
|
NM_139113 |
Nr2f6 |
nuclear receptor subfamily 2, group F, member 6 |
|
|
NM_012920 |
Camk2a |
Calcium/calmodulin-dependent protein kinase II alpha subunit |
|
|
NM_012806 |
Mapk10 |
Mitogen activated protein kinase 10 |
|
|
NM_214457 |
Phactr1 |
Phosphatase and actin regulator 1 |
|
|
NM_031707 |
Homer1 |
homer homolog 1 (Drosophila) |
|
|
NM_024378 |
Grid1 |
Glutamate receptor, ionotropic, delta 1 |
|
|
NM_017262/// NM_031508 |
Grik5 |
Glutamate receptor, ionotropic, kainate 5 |
|
|
NM_001004238 |
Rab21 |
RAB21, member RAS oncogene family |
|
|
NM_031836 |
Vegf |
Vascular endothelial growth factor A |
|
|
NM_017290 |
Atp2a2 |
ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 |
|
|
NM_012713 |
Prkcb1 |
Protein Kinase C, beta 1 |
|
|
NM_017041 |
Ppp3ca |
Protein phosphatase 3, catalytic subunit, alpha isoform |
|
|
NM_001033670 ///NM_00103361 ///NM_001033672 ///NM_031535 |
Bcl2l1 |
Bcl2-like 1 |
Tutti i geni regolati sono stati classificati in base al processo biologico nel quale sono coinvolti, secondo le modalità indicate dal Gene Ontology Consortium. Dall'analisi è emerso che la maggior parte di essi partecipa alla regolazione del metabolismo e della fisiologia cellulare (Tabella 9.)
TABELLA 9.
|
Gene Ontology: biological process |
Genes |
Nº genes |
Percentage |
|
1398822_at 1389195_at 1392180_at 1384594_at 1395102_at 1385592_at 1383554_at 1368924_at 1368146_at 1380548_at 1384061_at 1383672_at 1393378_at 1376247_at 1386995_at 1375043_at 1389686_at 1398862_at | |||
|
1389195_at 1392180_at 1384594_at 1385592_at 1383554_at 1368146_at 1384061_at 1383672_at 1393378_at 1376247_at 1386995_at 1375043_at 1389686_at 1398862_at | |||
|
1389195_at 1392180_at 1385592_at 1380548_at 1384061_at 1393378_at 1386995_at 1375043_at 1398862_at | |||
|
1389195_at 1392180_at 1385592_at 1368924_at 1380548_at 1384061_at 1393378_at 1386995_at 1375043_at |
|
||
|
1389195_at 1392180_at 1368924_at 1380548_at 1384061_at | |||
In particolare, 9 geni codificano proteine recettoriali o proteine implicate nei meccanismi di trasduzione del segnale (Grid1, Grik5, Gabrb2, Gna12, Gpr135, Mapk10, Homer1, Rab21, RABIN3), 6 geni codificano proteine coinvolte nell'omeostasi intracellulare del calcio (Atp2a2, Prkcb1, Camk2a, Ppp3ca, Plcb1, Prkce), 6 geni codificano proteine implicate nei processi di adesione cellulare (Nrcam, Eml2, Itga6, Cdh22, Cdh1, Phactr1), 5 nell'apoptosi (Dcc, Esrrbl1, Tieg, Ebag9, Bcl2l1), 1 nella risposta infiammatoria (Ptgs2) e 12 nella regolazione della trascrizione (Eif2b5, Nr4a1, Nr4a3, Nr2f6, Pou3f1, Egr1, Egr2, Cebpb, Nfx1, Cnot7, Fos, Junb).
Sulla base di tali risultati è stata avviata l'ultima fase del lavoro, mirata a confermare, mediante RT-PCR semiquantitativa e real-time PCR, i dati sperimentali ottenuti con la tecnica del DNA array. Sono stati scelti in via preliminare due gruppi di geni coinvolti nell'apoptosi e nei processi di adesione cellulare. I geni oggetto di studio coinvolti nell'apoptosi sono Bax, Bad, Bcl2, Bcl2l1, Tieg, Ebag9, Sgk e Stk17b, mentre quelli implicati nell'adesione cellulare sono Nlgn3, Itgam, Itga6, Hes5, Cldn10, Cdh1, Cdh7 e Cdh22.
Figura 7. Regolazione genica nella corteccia frontale del cervello di ratto in 3 condizioni sperimentali differenti. BAS=ratti naive trattati con salina CMS= ratti sottoposti a protocollo di stress cronico e trattati con salina TRATT= ratti sottoposti a protocollo di stress cronico e trattati con QTP 2 mg/kg/die A) Risultati Real time RT-PCR B) Risultati RT-PCR semiquantitativa C) Risultati DNA array. E' indicato il confronto tra i gruppi CMS/BAS e TRATT/CMS relativamente alla regolazione del gene in esame
A)
![]()
![]()
![]()
![]()

B) ù
BAS CMS TRATT
394
bp -
![]()
BAS CMS TRATT
422
bp -
![]()
BAS CMS TRATT
484
bp -
![]()
BAS CMS TRATT
233
bp -
![]()
BAS CMS TRATT
280
bp -
 Actina
Actina
C)
DNA array - ( -( - (
Figura 8. Regolazione genica nella corteccia frontale del cervello di ratto in 3 condizioni sperimentali differenti. BAS=ratti naive trattati con salina CMS= ratti sottoposti a protocollo di stress cronico e trattati con salina TRATT= ratti sottoposti a protocollo di stress cronico e trattati con QTP 2 mg/kg/die A) Risultati Real time RT-PCR B) Risultati RT-PCR semiquantitativa C) Risultati DNA array. E' indicato il confronto tra i gruppi CMS/BAS e TRATT/CMS relativamente alla regolazione del gene in esame
A)
![]()
![]()
![]()
![]()

B)
BAS CMS TRATT
284
bp -
![]()
BAS CMS TRATT
534
bp -
![]()
BAS CMS TRATT
519
bp -
![]()
BAS CMS TRATT
220
bp -
![]()
BAS CMS TRATT
280
bp -
![]() Actina
Actina
C)
DNA array - ( - ( ↑ (↓)
Figura 9. Regolazione genica nella corteccia frontale del cervello di ratto in 3 condizioni sperimentali differenti. BAS=ratti naive trattati con salina CMS= ratti sottoposti a protocollo di stress cronico e trattati con salina TRATT= ratti sottoposti a protocollo di stress cronico e trattati con QTP 2 mg/kg/die A) Risultati Real time RT-PCR B) Risultati RT-PCR semiquantitativa C) Risultati DNA array. E' indicato il confronto tra i gruppi CMS/BAS e TRATT/CMS relativamente alla regolazione del gene in esame
A) 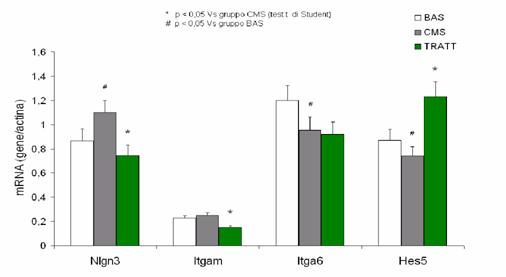
![]()
![]()
![]()
![]() B)
B)
BAS CMS TRATT
337
bp -
![]()
BAS CMS TRATT
324
bp -
![]()
BAS CMS TRATT
526
bp -
![]()
BAS CMS TRATT
174
bp -
![]()
BAS CMS TRATT
280 bp -
![]() Actina
Actina
C)
DNA array - ( - ( ( -
Figura 10. Regolazione genica nella corteccia frontale del cervello di ratto in 3 condizioni sperimentali differenti. BAS=ratti naive trattati con salina CMS= ratti sottoposti a protocollo di stress cronico e trattati con salina TRATT= ratti sottoposti a protocollo di stress cronico e trattati con QTP 2 mg/kg/die A) Risultati Real time RT-PCR B) Risultati RT-PCR semiquantitativa C) Risultati DNA array. E' indicato il confronto tra i gruppi CMS/BAS e TRATT/CMS relativamente alla regolazione del gene in esame
A)
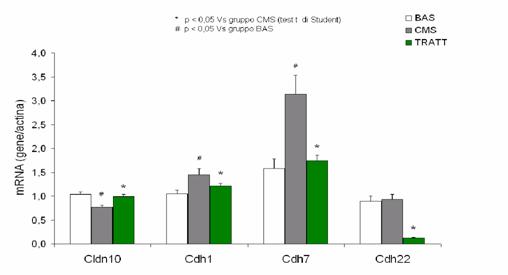
![]()
![]()
![]()
![]()
B)
BAS CMS TRATT
512
bp -
![]()
BAS CMS TRATT
274
bp -
![]()
BAS CMS TRATT
389
bp -
![]()
BAS CMS TRATT
753
bp -
![]()
BAS CMS TRATT
280
bp -
![]() Actina
Actina
C)
DNA array - ( - (
Alcuni recenti studi clinici hanno dimostrato che
In un precedente lavoro, Kapur e coll. (2003) hanno
voluto porre l'accento sulle differenze tra uomo e roditore per ciò che
riguarda la farmacocinetica della QTP, sottolineando come nel roditore
l'emivita degli antipsicotici atipici sia 4 - 6 volte più breve rispetto
all'uomo. Di conseguenza, secondo gli autori, gli studi su roditori effettuati
con protocolli di somministrazione cronica non sarebbero omologabili alla
condizione clinica, in quanto si raggiungerebbero livelli plasmatici troppo bassi di farmaco. Per ciò che riguarda
Come prevedibile, anche
Una prima importante conferma di questa ipotesi deriva dall'analisi dei risultati del DNA array, che ha consentito di valutare le modificazioni della regolazione genica nella corteccia frontale di ratti naive, anedonici e di ratti anedonici trattati con QTP. Apparentemente, sembrano infatti coinvolti nella comparsa dell'anedonia, uno dei sintomi cardine della depressione, alcuni geni che regolano l'apoptosi e i fenomeni di adesione cellulare, geni coinvolti nell'omeostasi del calcio intracellulare e numerosi fattori di trascrizione. Il DNA array è però una tecnica soggetta, come è noto, a possibili errori nell'interpretazione dei dati; risultati falsi positivi o falsi negativi sono probabili. Il protocollo CMS utilizzato in questo lavoro è sicuramente riconducibile ad una situazione di questo tipo, in quanto si basa sulla somministrazione quotidiana di stimoli molto blandi, anche se ripetuti per diverse settimane. E' prevedibile che queste condizioni, tutt'altro che estreme, provochino reazioni adattative appena rilevabili nel genoma delle cellule nervose, al contrario di quanto succede per esempio in seguito all'applicazione di uno stimolo stressogeno molto intenso come lo stress da costrizione (2-8 ore al giorno) ripetuto per 14 giorni (Kyoung-Shim e Pyung-Lim, 2006). La fase successiva del lavoro oggetto di questa tesi è stata perciò dedicata alla validazione dei risultati del DNA array mediante RT-PCR semiquantitativa e real time RT-PCR quantitativa. Sono stati scelti a tal proposito alcuni geni coinvolti nei processi apoptotici (Bax, Bad, Bcl2, Bcl2l1, Ebag9, Tieg, Sgk, Stk17b) e di adesione cellulare (Itga6, Itgam, Cdh1, Cdh7, Cdh22, Nlgn3, Hes5, Cldn10) regolati in modo eterogeneo (up- o down-regolati solo in un gruppo o in più gruppi) nel DNA array. Il confronto dei risultati ottenuti con le tre diverse metodiche indica una completa corrispondenza nella regolazione per Bcl2 e Stk17b (non regolati dallo stress, up-regolati da QTP), Tieg e Cdh7 (up-regolati dallo stress, down-regolati da QTP), Itgam (non regolato dallo stress, down-regolato da QTP), Itga6 (down-regolato dallo stress, non regolato da QTP). Una parziale correlazione tra i risultati è stata invece ottenuta per Bad, Cdh22 e Nlgn3 (down-regolati da QTP), Bax, Cldn10, Hes5 e Sgk (up-regolati da QTP) e Bcl2l1 (down-regolato dallo stress). Ebag9 e Cdh1 possono essere considerati falsi positivi in tutte le condizioni sperimentali prese in esame.
La famiglia di proteine Bcl2 regola l'apoptosi
controllando la permeabilità della membrana mitocondriale e il rilascio del
citocromo C. Le proteine anti-apoptotiche Bcl2 e Bcl-XL (prodotto del gene
Bcl2l1) risiedono sulla membrana esterna mitocondriale ed inibiscono la
liberazione del citocromo C dal mitocondrio. Le proteine pro-apoptotiche Bad,
Bid, Bax e Bim sono citosoliche e, in
seguito a segnali apoptotici, traslocano sui mitocondri dove promuovono il
passaggio del citocromo C nel citoplasma. Nel citosol il citocromo C lega Apaf1
e forma un complesso di attivazione con la caspasi 9, una proteasi a cisteina,
innescando il processo apoptotico. L'attività anti-apoptotica di Bcl2 è legata
alla formazione di omodimeri mediante il dominio N-terminale BH4. Bcl2 però può
essere parte anche di eterodimeri, in cui il secondo partner è spesso Bax. Il
legame di Bax con Bcl2 impedisce la formazione degli omodimeri. A sua volta Bax
può eterodimerizzare con altre proteine correlate a Bcl2. L'attività
anti-apoptotica, che risiede nella presenza di omodimeri di Bcl2, è quindi il
risultato di un delicato equilibrio di interazioni proteina/proteina in cui Bax
funge da ligando comune. Una riduzione dell'espressione di Bax o un aumento
dell'espressione di proteine correlate a Bcl2 che legano Bax, portano ad un
aumento della quota libera di Bcl2 e quindi a maggiore probabilità di
formazione di dimeri Bcl2 in grado di svolgere funzione anti-apoptotica. Per la
sopravvivenza delle cellule è perciò di vitale importanza il rapporto tra la
concentrazione di proteine pro- ed anti-apoptotiche. I nostri risultati per ciò
che riguarda il bilancio complessivo della regolazione dei geni coinvolti
nell'apoptosi, indica che lo stress cronico up-regola la trascrizione di Tieg
(pro-apoptotico) e riduce la trascrizione di Bcl2l1 (anti-apoptotico), ma non
ha effetto su Bcl2 e Bax.
Nel cervello adulto, una caratteristica peculiare del giro dentato dell'ippocampo è la neurogenesi la quale, associata all'apoptosi, conduce ad un rapido turnover delle cellule neuronali in questa regione del SNC. Le cellule in proliferazione migrano dalla zona subgranulare dell'ippocampo allo strato delle cellule granulari dove danno origine a neuroni maturi. La neurogenesi ippocampale è stata dimostrata in numerose specie animali, uomo compreso, ed è regolata da segnali endogeni ed esogeni di vario tipo. Per esempio, è stato dimostrato che l'esposizione ad uno stress acuto o ad uno stress cronico può sopprimere la proliferazione e la sopravvivenza delle cellule granulari del giro dentato (Czèh e coll. 2002; Pham e coll. 2003; Heine e coll. 2004).
Negli ultimi anni, numerosi autori hanno avanzato l'ipotesi che alterazioni della neurogenesi siano coinvolte nell' eziopatogenesi del DDM (Jacobs e coll. 2000; Eisch. 2002; Kempermann e Kronenberg, 2003). La dimostrazione che il trattamento cronico con litio e antidepressivi e la terapia elettroconvulsiva sono in grado di stimolare la neurogenesi nel giro dentato sembrano rafforzare questa ipotesi (Chen e coll. 2000; Malberg e coll. 2000; Scott e coll. 2000).
Tuttavia, il significato e l'importanza funzionale dei neuroni generati nell'ippocampo e il ruolo che l'arresto della plasticità neuronale nel giro dentato possano avere nei disturbi dell'umore, è ancora oggi oggetto di acceso dibattito nella comunità scientifica.
Studi di imaging cerebrale effettuati nell'uomo hanno anche rivelato che l'ippocampo può subire una riduzione del proprio volume in malattie stress-correlate come, ad esempio, il DDM (Sheline e coll. 2003). Uno studio analogo, effettuato nei primati, ha confermato che lo stress cronico provoca una riduzione delle dimensioni dell'ippocampo e che questo effetto è prevenuto dal trattamento con antidepressivi (Czèh e coll. 2001).
I meccanismi responsabili della perdita di volume dell'ippocampo non sono affatto chiari, dal momento che studi post-mortem effettuati in cervelli di pazienti affetti da DDM non hanno dimostrato una perdita netta di neuroni nell'area in questione. In più, nel DDM fenomeni apoptotici sono stati descritti solo in misura limitata in alcune regioni come la corteccia entorinale e il subiculum (Lucassen e coll. 2001; Muller e coll. 2001).
Nel complesso, gli studi precedentemente descritti non hanno chiarito il ruolo che la neurogenesi e la morte neuronale programmata possono rivestire nel danno provocato dallo stress nel cervello adulto e nella comparsa di malattie di interesse psichiatrico come i disturbi dell'umore.
E' certo che l'apoptosi e una perdita netta di neuroni sono dimostrabili nel tessuto cerebrale dell'animale da esperimento solo in situazioni di stress molto intenso e prolungato, che sicuramente non riproducono fedelmente le condizioni proprie della patologia umana. Al contrario, è probabile che nelle aree cerebrali coinvolte nella patogenesi dei disturbi affettivi (corteccia prefrontale, amigdala, ippocampo) si verifichino variazioni dei meccanismi regolatori dell'apoptosi, non sufficienti ad indurre direttamente la morte neuronale, ma capaci di alterare il delicato equilibrio intracellulare tra fattori pro- ed anti-apoptotici. I risultati ottenuti con il lavoro sperimentale oggetto di questa tesi sembrano in linea con questa ipotesi e possono rappresentare un iniziale contributo all'identificazione dei bersagli sensibili all'azione dello stress cronico (e dei farmaci stabilizzanti dell'umore come QTP) nei processi apoptotici.
Per quanto riguarda la regolazione dei geni
coinvolti nell'adesione cellulare, i risultati del DNA array indicano
complessivamente che lo stress cronico up-regola Cdh7, riduce la trascrizione
del gene Itga6, ma non ha effetto su Itgam.
Le caderine partecipano alla formazione delle
giunzioni aderenti, siti di interazione tra cellule, implicati non solo nella
formazione di parenchimi e superfici mucose, ma anche dell'organizzazione dei
contatti sinaptici. Esse si associano a formare omodimeri che interagiscono con
caderine dello stesso tipo espresse da cellule omologhe. Le interazioni mediate
dalle caderine sono strettamente dipendenti da ioni calcio che si legano alla
molecola. Nel versante intracellulare esse si associano ad altre proteine
(catenine) e al citoscheletro. Il complesso caderine-catenine-actina e altre
proteine citoscheletriche (α-actinina) permette l'ancoraggio delle
caderine nel loro versante citoplasmatico e funge da nucleo di aggregazione per
altre molecole di segnale che regolano diverse funzioni cellulari. Oltre che
interagire tra loro, le cellule aderiscono al tessuto interstiziale o a
membrane basali, strutture che nel loro insieme vengono considerate matrice
extracellulare. L'adesione alla matrice extracellulare è prevalentemente
mediata da integrine. Le integrine sono eterodimeri costituiti
dall'associazione non covalente tra una catena α e una catena ß e vengono
classificate in diverse sottofamiglie in base alla struttura della catena ß.
Membri delle sottofamiglie ß1, ß3, ß4, ß5,
ß6 e ß8 riconoscono proteine della matrice extracellulare
quali collagene, fibronectina, vitronectina, laminina ed entactina. In
letteratura, le notizie sulla regolazione e sulle funzioni specifiche di Cdh7,
la cui trascrizione è aumentata dallo stress cronico e riportata ai livelli
basali dal trattamento con QTP nelle nostre condizioni sperimentali, sono
scarse (Kawano e coll. 2002) e non consentono una interpretazione soddisfacente
dei risultati ottenuti. Le stesse considerazioni valgono per il gene Itga6,
down-regolato dallo stress cronico. Di tutt'altro interesse potrebbe essere
invece il dato relativo alla up-regolazione del gene Hes5 da parte della QTP.
La proteina Hes5 è un fattore di trascrizione che, assieme a NUDR, regola negativamente l'espressione del
recettore 5-HT1A serotoninergico. Il recettore 5-HT1A è un recettore somatodendritico con
funzioni inibitorie sul firing dei neuroni serotoninergici dei nuclei del rafe
ed è perciò considerato l'effettore della modulazione negativa delle vie
serotoninergiche nel SNC. La riduzione della funzione serotoninergica, come è
noto, è ritenuta in parte responsabile della patogenesi dei disturbi dell'umore
e un aumento della densità dei recettori 5-HT1A nei nuclei del rafe è
stato dimostrato in studi post-mortem effettuati sul cervello di pazienti
depressi (Stockmeier e coll. 1998). Inoltre, molti farmaci in grado di
contrastare i sintomi dell'ansia e della depressione, compresa
Centorrino F., Baldessarini R.J., Kando J., Frankenburg F.R., Volpicelli S.A., Puopolo P.R., et al.' 1994. Serum concentrations of clozapine and its major metabolites: effects of co-treatment with fluoxetine or valproate. Am. J. Psychiatry 151, 123-5.
Cheeta S., Broekkamp C., Willner P., 1994. Stereospecific reversal of stress-induced anhedonia by mianserin and its (+)-enantiomer. Psychopharmacology 116, 523-528.
Cheeta S.,
Cheeta, S., Ruigt, G., van Proosdij, J., Willner, P., 1997. Changes in sleep architecture following chronic mild stress. Biol. Psychiatry 41, 419-427.
Chen, G., Rajkowska, G., Du, F., Seraji-Bozorgzad, N., Manji, H.K., 2000. Enhancement of hippocampal neurogenesis by lithium. J. Neurochem. 75, 1729-1734.
Connor T.J. e al., 1999. Stressor-induced alterations in serotoninergic actyvity in animal model of depression. NeuroReport 10, 523-528.
Coryell, W.,Endicott, J., Keller, M., 1992. Rapidly cycling affective disorder: demographics, diagnosis, family history and course. Arch. Gen. Psychiatry 49, 126-131.
Gonzalez-Pinto A., Lalaguna B., Mosquera F., Perez de Heredia J.L., Gutierrez M. et al., 2001. Use of olanzapine in dysphoric mania. J. Affect. Disord. 66, 247-53.
Rang H.P., Dale M.M., Ritter J.M., Moore P.K., 2005. Farmacologia. 3a edizione, ed. Ambrosiana.
|
