Programma di ANATOMIA PATOLOGICA II
Parte sistematica, citologia, immunoistochimica
TUMORI DELLE PARTI MOLLI (Calì-Fiore-Donati)
Inquadramento classificativo
Tumori fibrosi
Tumori fibroistiocitari
Tumori lipomatosi
Tumori della muscolatura liscia e striata
Tumori vascolari (integrare il Sarcoma di Kaposi con gli appunti)
Tumori sinoviali
Tumori dei nervi periferici
Paragangliomi
Mesoteliomi benigni e maligni (integrare con gli appunti)
Disturbi di circolo delle ossa (Lanza)
Necrosi settiche e asettiche (Lanza)
Processi infiammatori specifici e aspecifici (Lanza)
Osteopatie disendocrine (paratiroidee, ipofisarie, ovariche, corticosurrenaliche) (Lanza)
Alterazioni ossee nel m. di Cooley (Lanza)
Osteomielosclerosi (Lanza)
Istiocitosi X (Lanza)
Cisti ossee (Lanza)
Tumori delle ossa (Ascenzi)
Mastiti (Lanza)
Mastopatia fibrocistica (Lanza)
Ginecomastia (Ascenzi)
Tumori (Ascenzi)
ENCEFALO (Ascenzi)
Malformazioni
Meningi: disturbi di circolo, flogosi, tumori primitivi e secondari
Encefaliti
Idrocefalo
Disturbi di circolo cerebrali
Tumori primitivi e secondari
OVAIO (Lanza)
Flogosi
Tumori
TUBA (Lanza)
Flogosi
Tumori
UTERO (Lanza)
Corpo dell'utero:
a) Iperplasia ghiandolare dell'endometrio
b) Endometriti
c) Endometriosi
d) Tumori
Collo dell'utero:
a) Cerviciti
b) Erosione della portio
c) Tumori
d) Displasie epiteliali dei collo uterino
Patologia connessa allo stato gravidico (Lanza)
a) Disturbi di circolo della placenta
b) Flogosi
c) Mola vescicolare
d) Corionepitelioma
e) Endometrite sinciziale
f) Gravidanza tubarica
RINOFARINGE (Ascenzi)
Tumori
LARINGE (Ascenzi)
Laringocele
Laringiti
Tumori
CAVO ORALE (Lanza)
Tumori
GHIANDOLE SALIVARI (Ascenzi)
Tumori
TIROIDE (Ascenzi)
Flogosi
Struma
Cisti (?)
Tumori
Correlazioni anatomo-cliniche nell'ipertiroidismo e nell'ipotiroidismo
PARATIROIDE (Ascenzi)
Iperplasie
Tumori
Quadri anatomo-clinici da iper ed ipofunzione
SURRENE (Ascenzi)
Corteccia surrenale
a) Alterazioni circolatorie
b) Flogosi (specifiche ed aspecifiche)
c) Iperplasie
d) Tumori
e) Quadri anatomo-clinici da iper ed ipofunzione
Midollare surrenale
a) Iperplasie
b) Tumori (serie simpatica e cromaffine)
IPOFISI (Ascenzi)
Alterazioni di circolo
Tumori
Correlazioni anatomo-cliniche
EPIFISI (Ascenzi)
Tumori
Quadri anatomo-clinici
PANCREAS ENDOCRINO (Lanza)
Tumori
SISTEMI ENDOCRINI DIFFUSI (Ascenzi)
Tumori
Precancerosi (Lanza)
Tumori (Lanza)
Nevi e melanomi (Ascenzi)
Citologia generale: esfoliativa, agoaspirativa e di apposizione.
Tecniche di prelievo, di allestimento e di fissazione.
L'identificazione del campione.
Tecniche di colorazione.
Criteri di malignità.
Modalità di refertazione.
Sensibilità, specificità, vantaggi e limiti della citologia.
Citologia polmonare: espettorato, brushing, liquido di lavaggio bronchiale, BAL.
Citologia urinaria
Citologia vaginale
Citologia del liquor
Citologia mammaria
Citologia dei versamenti
Citologia epatica
Citologia tiroidea
Quadri citologici dei tumori polmonari
Struttura e funzione di Antigeni e Anticorpi
Reazioni Antigene-Anticorpo
Metodi di Preservazione (chimici e fisici) dei Campioni per immunoistochimica
Reazioni dirette, indirette e a 3 stadi
Metodiche in immunofluorescenza: applicazioni, vantaggi e limiti
Metodiche immunoenzimatiche ed immunometalliche: applicazioni, vantaggi e limiti
Anticorpi policlonali e monoclonali
Applicazioni delle tecniche immunocitochimiche:
a) identificazione di microrganismi
b) antigeni oncofetali
c) proteine strutturali intracellulari
d) filamenti del citoscheletro
e) ormoni
f) recettori ormonali
g) indici proliferativi
h) fenotipizzazione linfocitaria
i) definizione di istogenesi e di differenziazione delle neoplasie
j) oncoproteine
k) diagnostica delle lesioni benigne e maligne della mammella
Nozioni applicative dì biologia molecolare:
a) tecniche estrattive e non estrattive
b) marcatura radioattiva e non radioattiva
c) Dot e Southern Blotting, Reazione Polimerasica a Catena
d) ibridazione in situ
e) applicazioni in diagnostica virologica, genetica ed oncologica
I vantaggi della citologia sono di far ottenere una diagnosi precoce e rapida, di poter essere alquanto indicativa (è possibile effettuare prelievi TAC-guidati), di essere ben accetta dal paziente, di essere semplice, ripetibile, economica; inoltre, è veloce nell'allestimento e, nelle lesioni di chiaro stampo benigno, evita la biopsia e allevia l'ansia. Nelle lesioni maligne, invece, consente l'effettuazione di indagini biologiche, cinetiche, recettoriali e di microscopia elettronica. La citologia, inoltre, [cito testualmente, NdR] consente un monitoraggio radiologico successivo, perché il nodulo è rimasto integro e inalterato.
Gli svantaggi, invece, sono dati dal fatto che viene offerto comunque un quadro limitato, c'è spesso necessità di conferma istologica, è possibile che il materiale sia inadeguato per la diagnosi (MID = Materiale Inadeguato per la Diagnosi), e si realizzano dei falsi positivi e falsi negativi.
I falsi negativi si realizzano per neoplasie molto differenziate oppure per lesioni minime (come ad es. quelle mammarie); i falsi positivi si realizzano per sopravvalutazione delle displasie. In questi casi bisogna rifiutare [cosa ???, NdR] oppure passare all'istologia.
La sensibilità è un indice della possibilità che un test ha di scovare la corrispondenza effettiva tra la percentuale di soggetti effettivamente malati e le positività; questa risulta calcolabile rapportando il numero di veri positivi alla somma di questi con i falsi negativi, e moltiplicando il tutto per 100. La specificità è invece la possibilità che il test ha di scovare la corrispondenza tra la percentuale di soggetti di fatto sani e le negatività; risulta calcolabile rapportando i veri negativi alla somma di questi ultimi con i falsi positivi, e moltiplicando il tutto per 100.
Morale finale = la citologia è un test molto specifico; la specificità della citologia è intorno al 99%, quindi se un cristiano risulta negativo, è probabile al 99% che sia effettivamente sano.
E' una colorazione tricromica, quindi utilizza tre coloranti. C'è un colorante nucleare, che è l'ematossilina alcolica di Harris (colora i nuclei in blu), e poi ci sono due coloranti citoplasmatici. Uno di questi due coloranti è sempre uno della serie Orange, e serve a colorare la cheratina; l'altro di questi sue coloranti appartiene alla serie EA, e serve per le sostanze acidofile. I coloranti EA infatti sono coloranti basofili. Ora, vi sono 6 tipi diversi di coloranti Orange, e ci sono 100 tipi di coloranti EA. I più usati, per questioni di standardizzazione, sono l'Orange n.6 (OG 6) e l'EA n. 50 (EA 50). Per gli amanti del masochismo, rivelerò che l'EA 50 è una soluzione alcolica di verde luce, bruno Bismarck e eosina.
La colorazione di Papanicolau è tra le più usate in citologia, ma non è l'unica. Altre colorazioni utilizzate sono:
a) PAS, per i mucopolisaccaridi;
b) PERLS, per i derivati emoglobinici [testualmente, NdR];
c) SOLODETZ, per i pigmenti Hbnici delle fibre d'asbesto;
d) GIEMSA, per ricerche ematologiche.
Sempre per accontentare i masochisti, diciamo che la colorazione di Giemsa è composta da blu di metilene, eosina, Azzurro II.
La gran parte dell'attività del laboratorio di Anatomia Patologica è oggi incentrata sulla citologia; il più delle volte c'è l'indicazione oncologica. La citologia si divide essenzialmente in citologia esfoliativa e citologia per aspirazione/con apposizione. Si tratta di due mondi molto diversi tra loro.
La citologia esfoliativa si avvale di campioni eliminati dal paziente oppure prelevati da organi comunicanti con l'esterno; e qui si possono fare una serie di esempi che però non facciamo: li vedremo volta per volta. La citologia per aspirazione/con apposizione invece prevede che i campioni vengano prelevati da organi non comunicanti con l'esterno. Anche in questo caso, vedremo in seguito le applicazioni; è inutile lambiccarsi il cervello per così poco.
Come viene acquisito il prelievo? Il prelievo va messo in un contenitore e debitamente identificato; si compila quindi un modulo di richiesta. Il fissativo che si utilizza in citologia è alcol, e va posto in pari quantità del materiale[2]. La consegna al laboratorio deve essere quanto più rapida possibile; in genere non si dovrebbero oltrepassare i 30 minuti.
C'è della gente che a cui piace descrivere il "modulo di richiesta" nei minimi particolari; in realtà io non lo farò, perché quel modulo lì vale solo per il Policlinico. A che pro farlo? E poi, non credo che una delle domande più gettonate all'esame sia "Mi parli del contenuto del terzo frame del modulo di richiesta; cosa c'è scritto al quarto rigo?".
Abbiam detto che il fissativo usato in citologia è alcol; in realtà, in alcuni casi si utilizza formalina, e questo vale i linfonodi, per le cellule epatiche, testicolari. Si tratta di cellule delicate, che l'alcol potrebbe rovinare. L'alcol, difatti, coarta gli elementi cellulari tirandovi fuori l'acqua molto rapidamente, mentre la formalina ha una azione più lenta.
Una volta che il campione è arrivato al laboratorio, si utilizza una citocentrifuga. Questo è fatto per meglio analizzare la parte corpuscolata. [Anche qui testualmente] C'è un filtro e un vetrino. L'apparecchio, ruotando, spinge il liquido verso il filtro[3]. Il liquido, giunto al vetrino, per forza centrifuga tende a disperdersi; il filtro assorbe il liquido e fa concentrare la cellule (il filtro ha un foro!). Il filtro è fatto di carta bibula. [Traduzione] L'apparecchio è composto da un filtro di carta bibula e da un vetrino, posto dopo di esso. Quando la centrifuga inizia a funzionare, il liquido viene spinto verso il filtro che, per la sua natura di filtro di carta bibula, trattiene il fluido e lascia passare le cellule, tanto più che il filtro possiede un foro centrale. Attraverso questo foro centrale cadono le cellule e si raccolgono sul vetrino sottostante.
Dopo che il citologo ha analizzato con varie tecniche il vetrino e ha così potuto soddisfare le sue turpi voglie, deve fare la refertazione. E qui, su apposto modulo (per il quale vale lo stesso ragionamento fatto prima per il modulo di richiesta), dovrà indicare se il materiale è valido o meno (MID), la provenienza del materiale, la quantità e qualità degli elementi osservati, un giudizio (positivo, negativo, sospetto, non diagnosticabile).
Alla fine, il clinico deve sapere, dall'esame citologico, se c'è una ripresa di malattia o meno e, se c'è, se il suo grado (granding) è uguale a prima oppure è superiore.
Questo è un paragrafo omaggio.
La citologia aspirativa viene effettuata sulla base di campioni che provengono da organi/cavità non comunicanti con l'esterno.
La citologia per apposizione invece sfrutta la superficie di taglio di linfonodi, milza, midollo osseo, encefalo. Prevede che dopo apposizione tissutale sul vetrino, si effettui una colorazione rapida con Giemsa. Il vantaggio maggiore è dato dalla rapidità di esecuzione; lo svantaggio è che vengono persi i rapporti strutturali.
In verità si tratta di due pratiche completamente diverse. Chissà perché sono state accomunate nella classificazione iniziale?
Utilizza un ago sottile, che è simile all'ago a farfalla; è collegato con un tubicino trasparente ad una siringa, entro la quale si raccoglie l'aspirato. La citologia aspirativa è utilizzata in diverse patologie.
Nelle neoplasie mammarie, se il nodulo è circoscritto; nelle neoplasie tiroidee, in ambito ospedaliero poiché c'è elevato rischio di emorragia (previa Ecografia!); nelle linfoadenopatie superficiali, se il linfonodo è ingrossato, dolente (flogosi, linfomi[4], metastasi); nelle neoplasie dei tessuti molli, che spesso sono forme miste; a proposito di quest'ultime neoplasie, è inutile cercare le mitosi, proprio per via della eterogeneità istologica.
Ancora, questo tipo di citologia è usata per le neoplasie polmonari periferiche, sebbene si tratti di una metodica molto indaginosa (prevede una guida TAC), per le neoplasie epatiche, spleniche (primitive o metastatiche), prostatiche (agoaspirato sotto giuda Eco o TAC), testicolari.
L'aspirazione può essere singola, cioè con un solo passaggio attraverso la cute, oppure multipla, cioè con punture in più punti.
Pensavate di sapere tutto sui criteri di malignità? Pensavate che ormai non c'erano più segreti tra voi e il fantastico mondo delle displasie? Bene, leggendo questo capitolo vi accorgerete che avevate ragione.
I criteri di malignità sono suddivisibili in segni indiretti, criteri di gruppi di cellule, criteri di singole cellule.
Segni indiretti di malignità. Sono rappresentati dall'abbondanza di cellule, dalla perdita della loro adesività (le cellule tendono a staccarsi), dalla presenza di materiale necrotico sul fondo del vetrino.
Criteri di malignità di gruppi di cellule. Cioè desunti osservando gruppi di cellule. Questi sono conferiti da particolari disposizioni spaziali delle cellule sul vetrino (ad es., perle cornee srotolate, che assumono un tipico andamento), da specializzazioni di membrana (assenti quando dovrebbero essere presenti, oppure il contrario), da una certa coesività reciproca.
La disposizione spaziale delle cellule è intuibile osservando la disposizione dei nuclei e dei citoplasmi; da questa è possibile capire se ci si trova davanti ad una struttura papillare (i nuclei delle cellule che costituiscono la papilla sono tutti in posizione centrale) o tubulare (i nuclei sono tutti basali, e la cellula ha un secreto apicale).
Le specializzazioni di membrana, come villi, ciglia, microvilli, sono un segno di differenziazione, ma anche un segno di "deriva" istologica. Ad es., che ci fa una cellula simil-epitelio bronchiale in utero?
Criteri di malignità di singole cellule. Cioè desunti osservando la singola cellula. Alcuni di questi criteri riguardano la cellula nella sua totalità, altri solo il citoplasma, altri ancora solo il nucleo.
La cellula nella sua totalità può mostrare alterazioni della struttura citoarchitettonica, di volume, di forma, alterazioni degenerative, necrotiche. E sono tutti segni di malignità.
Il citoplasma può mostrare una quota non normale di acidofilia (Orangiofilia[5]) o basofilia (EA), o vacuoli.
Il nucleo può mostrare un aumento di volume (per aumento di DNA e/o proteine e/o quota di idratazione); a questo proposito c'è da dire che un diametro nucleare superiore a 10 nm deve dare sospetto di neoplasia o di stimolo flogistico/ormonale. Ancora, altre caratteristiche nucleari maligne sono una alterazione del rapporto nucleo/citoplasma, la presenza di nuclei multipli, di nucleoli, di ipercromatismo nucleare (aumento di DNA), di cromocentri (segno di aumento dell'attività nucleare), la irregolarità della membrana nucleare, la presenza di mitosi atipiche (ad es., asimmetriche, o con fusi in numero aumentato, o senza fusi, o con aumento del numero di cromosomi, o con presenza di cromosomi aberranti).
Esame dell'espettorato. Innanzitutto le indicazioni, cioè "chi lo deve fare?". L'esame dell'espettorato è indicato per diagnosi microbiologica (ad es., ritrovamento del bacillo tubercolare) o, anche, oncologica. Viene condotta in pazienti, e quindi in soggetti clinicamente affetti, in soggetti in possibile stato preclinico e, altra possibilità, come fatto preventivo.
La prima cosa da chiarire è se quanto raccolto rappresenta effettivamente espettorato o trattasi di sputo. In realtà è facile accorgersene, dal momento che nello sputo non ci sono cellule delle basse vie respiratorie, e quindi i macrofagi alveolari. Ci sono dei criteri standardizzati che servono a differenziare i due campioni. L'espettorato da esaminare in citologia deve essere il primo del mattino, e per tre mattine di seguito. Per una buona espettorazione, si invita il paziente a collaborare facendolo sedere a letto a abbracciare un cuscino, così da permettere una massima espansione toracica e quindi favorire una tosse quanto più profonda.
L'espettorato va raccolto in un contenitore particolare, detto "scatola di Petri" e, se il laboratorio verrà raggiunto entro 30 minuti, va portato così com'è; in caso contrario occorrerà utilizzarvi alcol denaturato al 95%. La risposta si ha la mattina successiva al prelievo. Il laboratorio fornisce tutte e tre le risposte, quindi, quattro giorni dopo. Queste risposte sono individualizzate per ciascun campione fornito, che potrà essere positivo, negativo, MID. In caso di MID, bisogna ripetere, e si deve vagliare la possibilità di somministrare preventivamente dei fluidificanti.
Esame delle urine. Viene condotto nella prima gestione del paziente urologico, nei pazienti con ematuria e nei follow-up dei carcinomi vescicali. Per effetuare l'esame delle urine, si raccolgono le seconde urine (cioè non le prime della giornata), che hanno poco stazionato in vescica e quindi sono state meno soggette ad attacchi acidi denaturanti. Anche in questo caso, il materiale va inviato così com'è se il laboratorio è vicino, altrimenti si userà alcol 95%.
Dopo centrifugazione, il sedimento può essere strisciato sul vetrino oppure incluso e sezionato come un preparato istologico. Si valutano le caratteristiche delle cellule esfoliate dalla parete delle vie escretrici. La valutazione dei caratteri citologici sullo striscio colorato si avvale della distinzione in classi, secondo Papanicolau.
A questo punto dobbiamo fare una piccola digressione e parlare dell'urotelio. L'urotelio, 21321f53v cioè l'epitelio delle vie urinarie, è un epitelio di transizione, ed è formato da più strati; questa particolare disposizione è giustificata dalle funzioni che gli organi - cavi - che ne sono tappezzati (vescica, vie urinarie) rivestono, dovendosi estendere anche notevolmente. A vescica vuota osserviamo al microscopio uno strato di cellule basali, cubiche o cilindriche, più strati di cellule intermedie, poliedriche, e cellule superficiali ad ombrello. A vescica piena il tutto si distende, per cui si realizza uno scivolamento e appiattimento degli strati, con diminuzione del loro spessore e, nel contempo, aumento della superficie complessiva.
Non so perché, ma a questo punto deve essere ricordato che un carcinoma uroteliale in fase avanzata, cioè G3, tende alla desquamazione e a formare perle cornee.
Un'altra metodica utilizzata per lo studio delle cellule uroteliali è il lavaggio vescicale. Anche in questo caso, l'indicazione è per pazienti con ematuria. Si tratta di una indagine citologica tra le più invasive: si raccoglie il liquido di lavaggio vescicale dopo cateterizzazione dell'organo e 2-3 invii di soluzione fisiologica.
Esame del secreto mammario. In realtà ne parleremo più diffusamente in seguito, comunque si tratta di esaminare la famosa secrezione. Bisogna conoscere le caratteristiche della secrezione; sapere se, ad es., è ematica o sieroematica (quando è ematica è più grave), se è spontanea o provocata, se è monolaterale o bilaterale (se è bilaterale è dovuta a stimoli endocrini). Comunque sia, la secrezione viene raccolta sul vetrino e si striscia, allo scopo di ottenere un monostrato cellulare. Si fissa il tutto con alcol 95%, oppure con fissativo spray. Quando sia presente una lesione eczematosa del capezzolo, occorre, prima, effettuare una scarificazione locale. Nella lettura citologica del secreto mammario sono 'quanto mai' importanti caratteristiche come il fondo (pulito, sporco) e le caratteristiche di coesività cellulare.
Esame del secreto prostatico. Chi lo fa? I signori con disturbi della minzione e/o con emospermia, segno quest'ultimo di tumori prostatici o testicolari. Per raccogliere il secreto prostatico, la ghiandola va preparata con un massaggio prostatico per via rettale; quindi si raccoglie il secreto su 2-3 vetrini, si striscia e si fissa.
Esame del liquor. Il liquor cefalorachidiano va raccolto tramite puntura lombare a livello di L4-L5, in soggetti con meningite oppure nei bambini con meningosi leucemica. L'indicazione è, quindi, infettiva o neoplastica. Si prelevano quantità definite di liquor, e l'esame deve essere immediato, per via della delicatezza degli elementi in esame.
Un importante concetto è che in alcuni tumori vi possono essere degli elementi blastici che si nascondono in "santuari" (encefalo, ma anche testicoli) sfuggendo alla terapia; ciò è alla base delle recidive, che si svelano con la presenza di tali cellule nel liquor.
Tiroide.
La citologia della tiroide si avvale di un aspirato che deve essere prelevato
molto lentamente, per evitare lesioni vascolari. La tiroide è tra gli organi
più delicati. Per effettuare un aspirato tiroideo si usa lo stesso strumentario
che si usa in citologia mammaria (e che vedremo); si richiede una estrema
precisione topografica, per cui il paziente verrà seguito strumentalmente.
Quando non è possibile al citologo definire un "indizio citologico-diagnostico"
certo si parla si "neoformazione follicolare". Questo significa che bisogna
ripetere l'esame dopo un po'. Cioè, potrebbe trattarsi di un carcinoma
follicolare molto differenziato, oppure una struttura follicolare non
carcinomatosa. Più agevole, invece, la diagnosi relativa al carcinoma
papillare.
Le ghiandole e i loro prodotti, L'ipofisi, L'ipotalamo, La tiroide e le paratiroidi, Le ghiandole surrenali, Il pancreas, La ghiandola pineale
Altre
applicazioni della citologia. Si può esaminare in citologia uno
striscio orale e orofaringeo, su materiale ottenuto dopo scarificazione con una
spatola; oppure, un esame del lavaggio e spazzolato gastrico, ottenuto mediante
endoscopia (l'endoscopia consente anche di effettuare prelievi istologici per
biopsia). Altre possibilità sono l'esame della bile, dopo incannulamento della
papilla di Vater ed estrazione della bile (in tal caso l'esame è utile per
studiare le cellule dell'epitelio delle vie biliari) e lo striscio rettale, per
patologie localizzate o generalizzate [testualmente, NdR].
EPATOCARCINOMA - TUMORI VIE BILIARI
https://www.epertutti.com/biologia/EPATOCARCINOMA-TUMORI-VIE-BILI73858.php
E' lo studio citologico del secreto cervico-vaginale. La sua indicazione è oncologica, batteriologica e ormonale. Le modalità di studio sono leggermente diverse di volta in volta. L'esame si chiama anche PAP test. Si colora con Papanicolau.
Indicazione oncologica. E' molto importante che lo striscio vaginale venga effettuato per motivi di prevenzione; i tal caso sono invitate a sottoporsi a tale esame tutte le donne in età fertile. Sono stati stabiliti dei calendari preferenziali per cui da 18 a 35 anni occorrerebbe fare un controllo all'anno, e per età da 35 a 60 anni un controllo ogni 3 anni. E, comunque, i controlli dovrebbero essere effettuati a partire dal primo rapporto sessuale.
Il soggetto che deve sottoporsi all'esame deve essere ben preparato: deve evitare le irrigazioni vaginali (volgarmente dette "lavande") per 48 ore prima dell'esame, così come i rapporti sessuali. L'esame va compiuto tra il 10° e il 15° giorno del ciclo; in fase preovulatoria, infatti, c'è il massimo della leggibilità per via del fatto che c'è poco muco, le cellule sono facilmente staccabili, ed è minima la proliferazione batterica.
Il soggetto va sdraiato su lettino ginecologico; per effettuare il prelievo si usa uno speculum lubrificato, che rende reale una cavità virtuale, e una particolare spatola di legno. I prelievi da effettuare sono 3. Il primo prelievo proviene dal fornice vaginale posteriore, il secondo prelievo proviene dalla portio uterina, a livello della giunzione squamo-colonnare, mentre il terzo prelievo è condotto in sede dell'endocervice, utilizzando un lungo cotton-fiocc o uno spazzolino. Il primo e il secondo prelievo vanno posizionati sullo stesso vetrino ma agli estremi opposti, e quindi vengono strisciati, ad occupare metà vetrino per uno. Lo striscio deve avvenire in senso reciprocamente inverso. Il terzo prelievo, invece, si srotola su un secondo vetrino tutto per sé. Il tutto viene fissato con alcol 95% oppure con fissatore spay, e i vetrini vengono identificati con il nome del soggetto interessato.
Il trasporto può essere fatto in scatola di cartone - se il prelievo è stato fissato con fissatore spray - oppure in apposite "scatole di Koplik", con tappo a vite - se il prelievo è stato fissato con alcol 95%. Per il PAP test c'è un modulo di richiesta a sé stante, per il quale valgono le solite considerazioni.
Studio batteriologico. Il prelievo è effettuato dal fornice vaginale, e lo striscio è posizionato al centro del vetrino, a spirale. Su questo, si pone soluzione fisiologica e un vetrino coprioggetto; al microscopio si provvederà ad identificare la flora batterica. In questo modo possono anche essere ricercati dei virus, ma l'esame è condotto un po' diversamente: si chiama esame virus PAP.
Studio ormonale. Va condotto in assenza di segni di flogosi, che possono alterare il risultato. Con la valutazione ormonale è possibile studiare la avvenuta ovulazione e la responsività periferica cellulare agli ormoni e alle terapie. Il prelievo è condotto al fornice vaginale posteriore, che è la zona più responsiva agli ormoni.
In fin dei conti. Ma, in fin dei conti, cosa si osserva al PAP test? Cellule, ovviamente, ma come?
a) Elementi celulari pavimentosi, provenienti da vagina ed esocervice, di solito di competenza dello strato superficiale ed intermedio. Fisiologicamente non dovrebbero mai comparire cellule basali e parabasali, a mano che non siano osservazioni in bambine o in donne in menopausa.
b) Elementi cellulari cilindrici, provienti dall'endocervice.
c) Altre cellule, quali cellule endometriali (fisiologiche se il prelievo è effettuato ebtro i primi 10 giorni del ciclo), elementi istiocitari, neutrofili, emazie, cellule modificate per fatti flogistici o da eventuali virus.
Il risultato del PAP test è espresso in 5 classi di Papanicolau, che sono riassunte nella tabella che segue:
|
Classi del PAP test secondo Papanicolau |
||
|
I |
Assenza di cellule atipiche | |
|
II |
Cellule atipiche senza caratteri di malignità | |
|
III |
Cellule atipiche con caratteri sospetti ma non dimostrativi di malignità |
dubbio |
|
IV |
Scarse cellule atipiche fortemente sospette di malignità | |
|
V |
Numerose cellule atipiche con netti caratteri di malignità | |
Accanto al PAP test merita un cenno anche lo striscio endometriale, che esiste, ma è condotto raramente, più che altro in donne portatrici di dispositivi intrauterini anticoncezionali (IUD). In questi casi si esamina il materiale stratificato sull'IUD: la presenza di un copro estraneo può dar luogo a manifestazioni iperplastiche. Oppure, lo striscio endometriale può essere condotto in caso di metrorragia o menorragia (iperplasia endometriale adenomatosa atipica).
Si esegue con una speciale spatolina. Il prelievo è strisciato sul vetrino in monostrato, si fissa con spray o alcol 95%, e si colora con Papanicolau. Si ricopre con vetrino coprioggetto.
A livello[6] della ghiandola mammaria è possibile effettuare studi di citologia esfoliativa e/o di apposizione, andando a studiare le lesioni ulcerative cutanee e del capezzolo oppure le secrezioni. Fra un po' andremo a vedere un po' meglio lo studio delle secrezioni. Altri possibili studi citologici della ghiandola mammaria sono mediante citologia agoaspirativa senza o con stereotassi. La differenza è che nel primo caso si esamineranno lesioni palpabili, ad es., un liquido cistico, nel secondo caso, invece, si apprezzeranno lesioni non palpabili e pertanto invenibili solo grazie ad adeguato strumentario (Eco, TAC). Questi ultimi casi prevedono una tecnica, detta di citoassistenza, che poi vedremo.
Secreto. La secrezione mammaria è fisiologica in gravidanza, nel puerperio, nell'allattamento. La secrezione patologica può essere monolaterale o bilaterale. Una secrezione monolaterale farebbe sospettare condizioni displastiche o neoplastiche, una secrezione bilaterale depone per un più rassicurante (!) disordine nella gestione della prolattina.
Il secreto mammario va interpretato alla luce dell'anamnesi, cioè delle condizioni necessarie perché si possa manifestare. Va raccolta effettuando una leggera pressione sul capezzolo e ponendo il tutto su vetrino. Si striscia, e si fissa con spray o con alcol 95%. Va valutanto anche macroscopicamente, e in ciò è importante l'aspetto ematico! Ne abbiamo già parlato qualche pagina fa.
Citologia aspirativa. Abbiamo già detto che può essere condotta per lesioni palpabili o per lesioni non palpabili. Una lesione è palpabile quando ha un diametro minimo di circa 1 cm. La lesione palpabile va sottoposta alla cosiddetta "tripletta diagnostica", con esame clinico, radiologico e citologico. Il nodulo va esplorato tra il 1/3 interno e i 2/3 esterni, per evitare di cogliere la possibile necrosi centrale, specie se il nodulo ha dimensioni abbastanza ragguardevoli (un diametro superiore ai 2 cm). E' possibile effettuare punture multiple. L'aspirato viene depositato su più vetrini, strisciato e fissato con alcol 95%. La parete della cisti (se di cisti si trattava) va ricontrollata palpando nuovamente la sede e con una pneumocistografia (evidenziazione radiologica del contorno).
Non finisce qui! Il "materiale" serve, oltre che per l'esame citologico, anche per la ricerca di recettori, per indagini di microscopia elettronica, per la ricerca degli indici di proliferazione neoplastica.
E che succede per le lesioni non palpabili? E' intuibile che in programmi di screening possa essere scoperta una di esse, e allora, mediante tecnica stereotassica[7], si effettua un prelievo. In realtà non sempre il materiale prelevato è diagnostico. In tal caso per migliorare la possibilità diagnostica si ricorre alla cosiddetta citoassistenza. In pratica è l'analisi microscopica dei vetrini preparati in via "estemporanea", quindi subito dopo il prelievo, senza troppi complimenti. E' possibile valutare se il materiale è diagnostico oppure è un MID, e le condizioni di benignità o meno.
L'esame citologico polmonare può essere condotto mediante brushing, lavaggio bronchiale, BAL. BAL non è una parolaccia, ma sta per "Lavaggio Bronco Alveolare". Il brushing e il lavaggio bronchiale hanno indicazione oncologica, mentre il BAL, andando ad esplorare il polmone profondo serve anche a studiare le pneumopatie interstiziali, la TBC, la sarcoidosi, ma anche le neoplasia periferiche. Anzi, proprio per le neoplasie periferiche è forse un metodo migliore della biopsia (in realtà questa frase è giustificata solo dal fatto che c'è una minore morbilità rispetto alla biopsia).
Brushing. Cioè "spazzolamento". Viene effettuato mediante fibroscopia. Il fibroscopio evidenzia una lesione ulcerativo-erosiva? Bene, si fa il brush. Il brushing è condotto per lesioni circoscritte, in fibroscopia (tramite il canale operatore del fibroscopio); il prelievo è adagiato e srotolato su una serie di vetrini, e la spazzola utilizzata per il prelevo va lavata in alcol 50% per recuperare il materiale rimasto tra le spatole.
Lavaggio bronchiale. Viene effettuato a paziente ospedalizzato e può essere condotto dopo l'esame dell'espettorato. Anche questo viene fatto in fibroscopia; si immettono due boli di 50 cc di soluzione fisiologica, quindi si recupera il liquido in provettoni [testualmente, NdR] graduati. In base alla quantità di liquido rilevata si valuta lo stato delle vie bronchiali. Ad es., nell'asma il iquido recuperato è minore del liquido immesso, mentre nell'enfisema succede il contrario. Il materiale deve giungere in laboratorio a fresco oppure fissato con alcol 95% in quantità pari al sedimento (quindi va centrufigato!).
Brushing e lavaggio bronchiale possono far diagnosi di carcinoma broncogeno occulto.
BAL. Con questa tecnica si ottiene materiale fluido e corpuscolato dalle ultime diramazioni dell'albero bronchiale. Il BAL va analizzato in tempi brevi, e quindi non va fissato. Consente uno studio citologico, immunoistochimico e microbiologico, dunque si fanno diversi campioni, per il citologo, per l'immunologo, per il microbiologo. Dopo centrifugazione, il sovranatante contiene lipidi, proteine, virus (il sovranatante va al microbiologo), mentre il sedimento contiene macrofagi, linfociti, polimorfonucleati, cellule desquamate (il sedimento va in immunoistochimica per l'esamina dei linfociti).
Cosa fa il citologo con il BAL? Il BAL viene centrifugato perché è piuttosto torbido; al liquido si aggiunge una pari quantità di alcol 95% che va cambiato più volte nella settimana (periodo di incubazione); si attende questo tempo perché dalla sospensione si formi una massa, che è come una pallina[8] di muco ed epitelio. Questa "pallina" dall'anatomopatologo viene allargata come un campione istologico. Quindi prima che il BAL sia pronto devono passare 8-10 giorni. La pallina viene poi inclusa in paraffina e tagliata al microtomo. La fettina ottenuta viene stesa in acqua, raccolta sul vetrino, sparaffinata, colorata con ematossilina/eosina . Con la colorazione vengono evidenziati i "grumi", che sono indice di materiale stratificato che non è utile ma sprecato [testualmente, NdR].
Neoplasie polmonari periferiche. Sono dette periferiche perché insorgono lontane dall'albero tracheo-bronchiale, cioè non vi aggettano.
[a questo punto occorre integrare da qualche parte sul "come vi si accede", cioè con quale tecnica]
Una volta ottenuto il campione citologico si esamina, ed è possibile riconoscere il quadro citologico. Qui è tutto da imparare a memoria.
Carcinoma squamoso differenziato pleomorfismo; raramente ci sono perle cornee, srotolate. Le cellule mostrano cromatina in grosse zolle adese alla membrana nucleare; non ci sono nucleoli. Il citoplasma è orangiofilo (presenza di cheratina), ialino, con aspetti di ectoendoplasma (cioè con differenza nella colorazione del nucleo e della zona subito a ridosso del nucleo). Si riscontra materiale corneo anucleato nella superficie epiteliale.
Carcinoma squamoso scarsamente differenziato pleomorfismo; non ci sono perle cornee. La cromatina è disposta in grosse zolle, oppure è dispersa; vi sono nucleoli. Il citoplasma è basofilo, più raramente orangiofilo. Il nucleo è a volte eccentrico.
Microcitoma Per cellule che hanno un diametro inferiore o uguale a 12,5 micron si parla di forma linfocito-simili, per cellule che hanno un diametro superiore a 12,5 micron si parla di forme a cellule intermedie. Si osservano cellule linfocito-simili organizzate in "rosette", scarsa/nulla coesività, allineamento in filiere, nucleo rotondo o con lacune, con cromatina a spugna; talvolta, presenza di nucleoli. Scarso citoplasma con alone basofilo.
Adenocarcinoma Si riscontrano cellule isolate o in cumuli; nucleo vescicoloso eccentrico, a cromatina dispersa. Il citoplasma possiede muco (raccolto in macronucleoli), e una vacuolizzazione più o meno grossolana.
Altre forme In realtà possono osservarsi anche forme di cancro polmonare che derivano dalla combinazione di più istotipi; ad es., microcitoma + carcinoma squamoso.
Qualcosa sulla pleura. La pleura ha origine mesoteliale. La cellula mesoteliale può evolvere in forme epiteliali e forme connettivali. La cellula mesoteliale normale ha un contorno nucleare normale, cromatina dispersa, più nucleoli. La cellula mesoteliale attivata ha un nucleo mitotico, si dispone a filiere e si sfalda in papille. La cellula mesoteliale attivata, invenibile in studi citologici, va studiata in tempi brevi, e non va usato alcol!
Il liquido pleurico contiene cellule rotonde con nucleo centrale e citoplasma basofilo. La morfometria aiuta a distinguere la provenienza cellulare quando nel liquido pleurico siano osservate cellule non tipiche. Il liquido pleurico va prelevato con puntura a livello del 6°-7° spazio intercostale sulla linea ascellare posteriore, o a livello del 6°-7°-8° spazio intercostale sulla linea angolo-scapolare. E' utile anche una valutazione chimica della composizione del liquido pleurico: la presenza di acido jaluronico in elevata concentrazione è fortemente indicativa di mesotelioma!
Bene, adesso scappo: devo comprare il fissatore spray.
Pubblicità della citologia
Colorazione di Papanicolau
Introduzione alla citologia
Piccola precisazione
Citologia aspirativa
Criteri di malignità
Tanti piccoli argomentini
Striscio vaginale
Citologia della ghiandola mammaria
Citologia polmonare (è l'ultimo)
Le tecniche utilizzate in immunoistochimica individuano le caratteristiche antigeniche, strutturali e funzionali di cellule e tessuti, utilizzando anticorpi marcati. Questi anticorpi sono da intendere in senso "laboratoristico" e non in senso biologico. Ad es., una delle tecniche in immunoistochimica è la ricerca di "antigeni" oncofetali. Si tratta di molecole espresse da alcune cellule nel periodo fetale e che, di regola, non vengono espresse nell'adulto, se non in alcune occasioni. Ora, il fatto che nell'adulto queste sostanze si chiamino "antigeni" in realtà non implica il fatto che verso di esse l'organismo formi degli "anticorpi". Anzi, mai sia! Sarebbe una reazione autoimmune. Tuttavia queste molecole sono chiamate antigeni dal momento che esistono delle immunoglobuline, usate in laboratorio (non umane!), con i quali antigeni reagiscono.
Esiste una scala antigenica; cioè la natura chimica di una molecola è in grado di essere più o meno immunogena. E quindi, le sostanze più immunogene di tutte sono le proteine; seguono DNA, glicidi e, da ultimi, e pertanto dotati di scarso potere immunogeno, i lipidi.
Per poter effettuare studi di immunoistochimica è necessario che il tessuto sia antigenico, quindi quest'ultimo deve essere trattato con criopreservazione (a basse temperature) e non con la formalina, che è denaturante! Altro concetto generale, ovvio e scontato, è che la sensibilità del legame Ag-Ig dipende dal numero di epitopi dell'antigene che legano l'anticorpo.
[qui manca una pagina nella dispensa originale]
Le metodiche usate in immunoistochimica sono:
a) Tecniche di immunofluorescenza
b) Tecniche immunoenzimatiche
c) Tecniche immunometalliche
Tutte queste tecniche sono accomunate dal fatto di usare anticorpi, che si legano agli antigeni rispettivi. Per andare a localizzare l'anticorpo legato, e quindi avere testimonianza del legame avvenuto, però, occorre "marcare" l'anticorpo prima che si leghi. E quindi, se il "marker" anticorpale è fluorescente, siamo nella tecnica a), se il "marker" è un enzima siamo nella tecnica b), se il "marker" è un metallo, siamo nella tecnica c).
E' chiaro che nel primo caso il marker evidenzierà "di suo" il legame avvenuto - e questo è evidenziabile con l'uso di un microscopio a fluorescenza -, mentre nel caso il cui il marker sia un enzima, occorrerà, per verificarne la presenza, far in modo che questo funzioni, e quindi le metodiche b) sono un po' più indaginose delle a); le tecniche c), invece, fanno uso di metalli pesanti che precipitano, e si manifestano come granuli elettron-densi opachi, evidenziabili in microscopia elettronica.
Immunofluorescenza. Può essere diretta o indiretta. La immunofluorescenza diretta prevede che ad un Ag cellulare (da svelare, pertanto) si leghi una Ig marcata con una sostanza fluorescente. Quindi tra l'Ag e l'Ig marcata non si interpone nulla. Ecco perché la metodica è detta diretta.
Tecnicamente, al preparato cellulare si aggiunge un siero con Ig marcate, quindi si procede a "lavaggio", causando così l'allontanamento di quanto non si è legato. E' chiaro che il lavaggio porterà via le Ig non legate, mentre quelle eventualmente legate non saranno portate via. E come facciamo ad accorgerci che alcune Ig sono rimaste lì, e quindi si sono legate? Osservando il preparato al microscopio a fluorescenza: le Ig aggiunte sono marcate e quindi, se si vede una fluorescenza, vuol dire che quelle Ig si sono legate, e il loro Ag è presente. Ora, la metodica del lavaggio viene effettuata sempre, prima della "prova del 9" dell'osservazione che prova l'avvenuto legame. Cioè, voglio dire che si fa sempre, in tutte le tecniche di immunoistochimica - e quindi non solo per l'IF diretta -, dopo aver fatto reagire il siero con le Ig con il preparato i cui Ag sono da svelare.
La immunofluorescenza diretta è rapida, ha un basso costo, è semplice, è ideale per malattie autoimmuni; però ha poca sensibilità, dal momento che, se l'Ag è presente in bassa quantità, il segnale emesso è basso, e quindi può non essere riconosciuto.
La immunofluorescenza indiretta, invece, prevede che all'Ag cellulare si leghi una Ig non marcata, alla quale poi si leghi una seconda Ig, stavolta marcata. Siccome alla prima Ig possono legarsi più Ig marcate, il risultato è amplificato, e quindi aumenta la sensibilità. Di solito la prima Ig (quella non marcata) è di topo, la seconda (quella marcata) è di coniglio. La metodica indiretta, quindi è più sensibile di quella diretta, consente una amplificazione del segnale, è ideale per sospensioni cellulari e per l'analisi quantitativa con tecniche automatiche; però ha lo svantaggio, comune peraltro a tutte le tecniche di immunofluorescenza, di avere un rapido decadimento, per cui l'opeatore deve fotografare il quadro osservato, altrimenti. addio.
Da qui parte la considerazione che vi sono svantaggi comuni alle tecniche di IF; esse sono la mancanza di dettaglio morfologico (difatti il vetrino è irriconoscibile), il fatto che vi è un'autofluorescenza tissutale che disturba e confonde (ad es., le fibre collagene), la necessità di operare in una stanza buia, l'operatore deve avere un occhio adattato, e la necessità di fotografare. Comunque, la IF è utile nelle patologie renali (svela i famosi "depositi") e nelle dermopatie (svela la presenza di immunocomplessi).
Citofluorimetria. Può essere statica o a flusso. L'apparecchio è in grado di scartare le cellule non marcate con l'Ig che abbiamo utilizzato (ad es., Ig anti-CD4); consente dosaggi anche quantitativi. In che consiste? In pratica è una macchina che analizza singolarmente le singole cellule e riconosce quelle alle quali vi è legata l'Ig. Nella citofluorimetria a flusso le cellule vengono fatte passare (il "flusso") davanti ad un sensore.
Immunoenzimatica. Prevede la presenza dell'Ag da ricercare, di una Ig marcata con un enzima e del substrato dell'enzima. Il prodotto della reazione enzimatica è opaco agli elettroni e permette la localizzazione intracellulare dell'Ag in esame. In realtà con tecniche particolari è possibile visualizzarne la presenza anche al microscopio ottico, e ciò è un punto di forza di questa metodica. Altri vantaggi sono una stabilità maggiore rispetto alla IF, la possibilità di dosaggi quantitativi (spettrofluorimetria). Però, vi possono essere dei tessuti che possiedono di per sé l'enzima marcante l'Ig: è chiaro che questi casi danno luogo a risultati falsi positivi. Perciò occorre inibire la attività enzimatica endogena; ad es., per eliminare la perossidasi bisogna incubare con H2O2.
Anche in questa tecnica
vi è una variante diretta (Ig marcato con l'enzima direttamente legato all'Ag
da ricercare) e una indiretta (cioè con una Ig "per lo mezzo"), come visto per
l'IF. La metodica indiretta più frequentemente utilizzata è la PAP, cioè Perossidasi Anti Perossidasi. In cosa
consiste? Come al solito, c'è l'Ag da dimostrare. A questo Ag viene legata una
Ig specifica, non 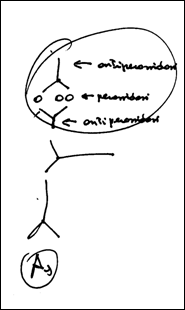 marcata. Questo complesso viene fatto
reagire con una seconda Ig, anch'essa non marcata, legante la prima Ig, e che
deve essere in concentrazione tale da creare un eccesso di Ig. In questo modo
potrà legarsi con uno dei due F(ab) all'Ig legata all'Ag, e avere
l'altro F(ab) libero, così come si vede in figura. A questo punto
viene aggiunto un sistema preformato detto PAP, cioè un complesso formato da 3
molecole di perossidasi (che è un enzima) e 2 Ig anti-perossidasi. Le Ig
anti-perossidasi devono legarsi alla seconda Ig utilizzata, come da figura. E'
importante che il complesso PAP abbia un rapporto stechiometrico di 3:2, in
modo tale che i F(ab) dell'antiperossidasi siano stabili e non
reagiscano. Altra nota è che se il I° Ig è di topo, anche il PAP deve essere di
topo, così che l'Fc dell' Anti Perossidasi possa legare il F(ab)
del II° Ig.
marcata. Questo complesso viene fatto
reagire con una seconda Ig, anch'essa non marcata, legante la prima Ig, e che
deve essere in concentrazione tale da creare un eccesso di Ig. In questo modo
potrà legarsi con uno dei due F(ab) all'Ig legata all'Ag, e avere
l'altro F(ab) libero, così come si vede in figura. A questo punto
viene aggiunto un sistema preformato detto PAP, cioè un complesso formato da 3
molecole di perossidasi (che è un enzima) e 2 Ig anti-perossidasi. Le Ig
anti-perossidasi devono legarsi alla seconda Ig utilizzata, come da figura. E'
importante che il complesso PAP abbia un rapporto stechiometrico di 3:2, in
modo tale che i F(ab) dell'antiperossidasi siano stabili e non
reagiscano. Altra nota è che se il I° Ig è di topo, anche il PAP deve essere di
topo, così che l'Fc dell' Anti Perossidasi possa legare il F(ab)
del II° Ig.
A che pro tutto questo? Per amplificare la presenza dell'Ag, che verrà svelata da ben 3 reazioni enzimatiche (le perossidasi sono 3).
Vi possono essere falsi positivi se l' Fc dell'antiperossidasi può aspecificamente fissarsi ad un tessuto.
Un'altra possibilità è di utilizzare il sistema avidina-biotina-perossidasi. Di cosa si tratta? E' una specie di sistema PAP, però più "in grande". Anche qui c'è un Ag, al quale viene fatta legare una Ig. A questa Ig viene legato successivamente un "sistema", composto da una Ig con 4 molecole di avidina; ad ogni molecola di avidina sono legate 4 molecole di perossidasi. Quindi ci sono, per ogni Ag, 16 molecole di perossidasi, e quindi di enzima. E la biotina a che serve?
In realtà la faccenda è un po' più complicata. All'Ag è legata una prima Ig. A questa prima Ig è legata una seconda Ig, che è biotinilata, cioè si tratta di una Ig che porta legata una molecola di biotina. A questo complesso viene successivamente legato un sistema molecolare composto da 3 biotine + 4 avidine; ad ogni avidina è poi legata una perossidasi. Finchè ad ogni avidina non sarà legata una biotina, non potrà avvenire la reazione della perossidasi. Quindi il sistema molecolare che noi andiamo ad aggiungere non funziona se l'unica avidina senza biotina non trova la biotina "utile". Se, dopo aver fatto avvenire tutti questi legami, la reazione della perossidasi avviene, allora significa che:
a) il complesso avidina-biotina-perossidasi si è completato;
b) il complesso avidina-biotina-perossidasi ha trovato la biotina mancante sulla Ig II°;
c) la Ig II° si è legata alla Ig I°;
d) la Ig I° si è legata all'Ag in ricerca;
e) l'Ag da ricercare esiste di fatto.
Più chiaro di così non so essere.
Nel presente contesto è opportuno descrivere la metodica della preparazione degli anticorpi monoclonali, che si fonda sulla reazione tra antigeni purificati e cellule immunocompetenti, dalle quali si desiderano ottenere anticorpi specifici.
La procedura si basa sulla formazione di un ibrido cellulare derivante dalla fusione di linfociti splenici di topi immunizzati con un determinato antigene e cellule mielomatose. Queste ultime sono dotate delle seguenti caratteristiche:
- hanno la capacità di replicarsi in vitro all'infinito in qualità di cellule neoplastiche;
- hanno perso la capacità di secernere anticorpi;
- non possiedono attività ipoxantina-fosforibosil-transferasica (HPRT).
Le cellule spleniche immunizzate vengono messe a contatto con le cellule mielomatose ed in presenza di glicol-polietilenico alcune di esse si fondono, dando luogo a degli ibridi cellulari. A fusione avvenuta gli ibridi vengono coltivati in terreni contenenti ipoxantina, aminopterina e timidina (HAT). Poiché le cellule mielomatose sono HPRT-deficienti muoiono in quanto non sono in grado di utilizzare la ipoxantina esogena per la produzione di purine. Infatti, la aminopterina dei terreni HAT blocca la sintesi di purine e pirimidine endogene.
Continuando a propagare le colture si finirà con l'ottenere ibridi che hanno ereditato l'immortalità della cellula mielomatosa ma sono in grado di sopravvivere nel terreno HAT. Gli ibridi vengono clonati in singoli pozzetti di una micropiastra ed i sovranatanti contenenti gli anticorpi monoclonali vengono saggiati (con metodiche immunoenzimatiche, ad esempio) per determinare la specificità verso l'antigene di partenza. I pozzetti contenenti gli ibridi desiderati vengono ad essere propagati in vitro o in vivo sotto forma di tumore ascite nei topi.
Dunque, esistono degli "anticorpi monoclonali" che reagiscono verso un unico antigene, e sono tutti uguali tra loro, cioè hanno tutti quanti la stessa specificità verso lo stesso epitopo dello stesso antigene. Gli anticorpi monoclonali sono estremamente specifici.
Al contrario, gli anticorpi "normali", cioè "policlonali", cioè diretti verso lo stesso antigene, ma ad epitopi diversi, sono meno specifici, in quanto può darsi che qualcuno di essi possa reagire verso epitopi identici presenti in altri Ag; tuttavia sono più sensibili rispetto ai monoclonali, dal momento che è possibile che più Ig leghino lo stesso Ag, avendo ogni Ag più epitopi.
E quindi, da tutto ciò si evince come la tecnica immunoistochimica sia effettivamente utile. In effetti, si tratta di tecniche che riescono ad identificare e a catalogare elementi tissutali per quello che in effetti rappresentano, al di là della morfologia. Ad es., come altrimenti differenziare leucemie per altri versi simili, se non con la determinazione dei CD di membrana? E la stessa cosa vale per i melanomi, che possono mimare qualsiasi tipo istologico, oppure per i carcinomi polmonari. In alcuni casi, inoltre, il laboratorio di immunoistochimica è di valido aiuto, anche perché la esecuzione e la lettura dei test è molto facile: è quanto avviene per la tiroide (con la ricerca dei recettori per la tiroglobulina), per la prostata (PSA), per la mammella. A proposito della mammella, è possibile individuare sulle cellule mammarie due proteine, HMFG II[10] e GCDFP ; queste sono "markers" importanti e utili per i casi metastatici.
Recettori ormonali. Si valuta se le cellule possiedono recettori per gli ormoni steroidei (i recettori per gli ormoni steroidei sono endocellulari). Questo è particolarmente utile in quei tumori che possono presentarsi in forma ormono-dipendente, come il tumore di mammella, prostata, ovaio (epiteliale), utero. Si tratta di un importante fattore prognostico: se il tumore è ormono-dipendente, è possibile impostare una terapia che, sfruttando tale caratteristica, ne blocchi l'attività. Diciamo che, in genere, quanto più alta è la differenziazione del tumore, tanto più probabile è l'ormono-dipendenza.
Oncogèni e oncoproteine. Sappiamo la storiella degli oncogeni e degli antioncogeni. Si tratta di geni cellulari la cui espressione rispettivamente esaltata o diminuita è alla base della manifestazione cancerosa. Le proteine codificate dagli oncogeni sono le oncoproteine. E' possibile, con le tecniche di immunoistochimica andare a valutare la loro presenza.
Ad esempio, nel retinoblastoma vi è una mutazione omozigote nel gene RB1, che è un gene oncosoppressore; nel neuroblastoma è amplificato l'oncogene MYC; nel CR mammario si realizza una mutazione del gene ERB B1, che diviene ERB B2. La presenza di ERB B2 dà luogo a recettori alterati che sono sempre attivati, con il risultato che la crescita cellulare è illimitata.
Antigeni oncofetali. Abbiam già detto di che si tratta. Assolutamente aspecifici - i loro livelli possono aumentare, oltre che in caso di neoplasia, anche in caso di febbre! - per la diagnosi, sono invece utili per il monitoraggio della terapia. Ad es., CEA, che è uno di essi, è un Ag oncofetale i cui livelli possono essere notevolmente aumentati in caso di cr colo-rettale; dopo trattamento, i livelli dovrebbero scendere e mantenersi bassi. Se aumentano nuovamente, significa che "qualcosa è cambiato". Altro Ag oncofetale di solito monitorato è l'-FP, che aumenta in caso di neoplasia epatica (ma non solo). Ce ne sono altri.
Ormoni. Possono essere studiati in immunoistochimica gli ormoni veri e propri prodotti dalle cellule. Per differenziare i tumori del pancreas endocrino, ad es., si ricorre alla ricerca di insulina, somatostatina, glucagone, gastrina. Sostanza P. Nel cr tiroideo si ricerca la tireoglobulina e la calcitonina. Nelle sindromi paraneoplastiche può essere utile verificare la produzione ectopica di ACTH, ADH, GH, come può avvenire nei tumori polmonari; idem per i carcinoidi.
Filamenti intermedi. Costituiscono la matrice di sostegno di tutte le cellule dell'organismo. Vengono detti "intermedi" a causa delle loro dimensioni, che sono intermedie tra i microfilamenti (per definizione con diametro fino a 6 nm) e i microtubuli (per definizione con diametro compreso tra 100 e 200 nm). I filamenti intermedi hanno caratteristiche e nomi diversi a seconda di dove ci troviamo. Per cui negli epiteli c'è la citocheratina, nel muscolo la desmina, nel tessuto mesenchimale la vimentina, nella glia c'è la proteina acida gliofibrillare, nel tessuto nervoso i neurofilamenti. In realtà le cose non sono così cristallizzate. Se la cellula cambia funzione, anche la qualità dei filamenti intermedi può variare; tuttavia, si tratta di situazioni che di solito non si verificano. Ad esempio, cellule epiteliali in coltura, e quindi disaggregate, esprimono anche la vimentina. La stessa cosa avviene per le cellule delle ghiandole salivari quando ci si trovi in presenza di un adenoma pleiomorfo.
Proteine strutturali. Si tratta di proteine, alcune delle quali hanno funzione contrattile. Le cellule della muscolatura liscia, striata e le cellule mioepiteliali esprimono actina e miosina. Le cellule della muscolatura striata esprimono anche mioglobina. Le cellule gliali, gli adipociti e la cartilagine esprimono la proteina S-100. Anche la ricerca immunoistochimica di queste proteine può essere utile.
Neoplasie mesenchimali. Le cellule mesenchimali sono rappresentate da cellule muscolari (liscie o striate), cartilaginee, ossee, adipose, fibrose, mixoidi, vascolari, istiocitarie, di Schwann. Sono tutte cellule vimentina +.
Le cellule istiocitiche sono anche + alla ricerca di altre proteine, come 1-AT (antitripisina), 1-ACT (antichimotripsina), CD68; in realtà CD68 non è sempre presente. Tutte le cellule istiocitarie - e mesenchimali in generale - hanno capacità fagocitarie in coltura. Attenzione: trovare 1-AT e 1-ACT potrebbe non significare "produzione propria", cioè da parte di cellule, ma potrebbe anche significare un inglobamento di tali molecole, esterne, all'interno delle cellule.
Le cellule di Schwann sono + alla proteina S-100, CD57, MBP ("proteina basica della mielina"), le fibrocellule muscolari lisce sono + a desmina, miosina, -ASMA (cioè actina), mentre le cellule della muscolatura striata sono + a desmina, miosina, actina, mioglobina. A proposito di queste ultime cellule, è allo studio la significatività di un gene e di una proteina "markers".
Le cellule vascolari (quelle che sono alla base, ad es., del sarcoma di Kaposi) sono + a vWF, CD31, CD34; le cellule ossee sono + a osteocalcina, osteotensina, vimentina, mentre i condrociti sono + a vimentina e osteotensina. I lipociti sono + alla proteina S-100; bisogna tuttavia considerare che il materiale lipidico prodotto è normalmente perso, con le comuni tecniche di preparazione.
I linfomi invece sono composti da cellule che hanno diverse positività a seconda della loro origine. Da qui la possibilità di classificare le forme linfomatose in svariati modi, e di indicarle, comunque, in base anche alle loro positività. Per le forme B-derivate, ad es., è possibile trovare le + per CD19, CD10, CD79A, Ig cellulari e di membrana. CD19 si riscontra solo se il campione è stato in precedenza congelato, mentre CD10 e CD79A si preservano anche con l'utilizzo di fissazione e inclusione. Le forme a derivazione T, invece, come il morbo di Hodgkin, hanno + a CD19, CD57, CD30, EMA. Le forme a derivazione macrofagica, poi, sono + a 1-AT, 1-ACT, CD68.
Ghiandola mammaria. E' una ghiandola tubulo-acinosa ramificata. Hanno funzione escretrice i duttuli e i dotti, mentre hanno funzione secretoria i lobuli ghiandolari. Le unità secernenti subiscono l'influenza ciclica di estrogeni, progesterone e, a termine della gravidanza, anche della prolattina. La componente secretoria, insieme alla porzione terminale del sistema duttale, è quella da cui origina la gran parte delle neoplasie mammarie. Essa è costituita da uno strato periluminale di cellule epiteliali, e da uno strato periferico. Lo strato periluminale ha una funzione secretoria; possiede citocheratina 19, e dimostra eosinofilia. Lo strato periferico, invece, è a contatto con la membrana basale, ha funzione contrattile (cellule mioepitelioidi), contiene actina, presenta una minore eosinofilia. Nelle neoplasie benigne ci sono cellule epiteliali e cellule mioepiteliali, in quelle maligne, invece, sono presenti tutte quante cellule epiteliali.
A livello di tali strutture è possibile, in immunoistochimica, fare il dosaggio in situ dei recettori ormonali, determinare oncogeni e oncoproteine, e determinare la quota di cellule proliferanti.
Determinazione della quota di cellule proliferanti. Viene effettuata tramite studi con timidina tritiata[12] (il cosiddetto "labelling index"), o con studi che utilizzino la bromodesossiuridina, o con l'indice mitotico oppure con la determinazione di alcuni "markers" particolari come PCNA o Ki67.
Il labelling index è il risulato di una tecnica che permette di determinare la quota di cellule proliferanti. In cosa consiste questa tecnica? Le cellule da studiare si mettono in coltura; nel mezzo di coltura c'è la timidina dove, al posto del normale idrogeno 1H, c'è il tritio 3H, isotopo. Quindi la timidina è tritiata. Le cellule, duplicandosi, devono sintetizzare nuovo DNA, e hanno bisogno di precursori. Questi precursori vengono presi dal mezzo di coltura; quindi, per ogni nuova cellule che si formerà, verrà consumata timidina tritiata. Il nuovo DNA che si formerà avrà timidina tritiata. Dopo un certo intervallo di tempo le cellule vengono fissate, disposte su un supporto particolare, e quindi si procede ad autoradiografia, cioè si pone, al di sopra del preparato, una pellicola ottica, che si impressioni in corrispondenza dei punti di presenza del tritio. Il grado di impressione della pellicola, quindi, è proporzionale alla presenza di tritio nelle cellule e quindi alla loro possibilità di proliferazione. Questa tecnica, concettualmente semplice, pone dei problemi sul piano pratico: difatti occorre tessuto vitale, c'è necessità di avere grandi campioni, c'è il problema della radioattività, e inoltre il tritio ha una breve emivita.
Con la bromodesossiuridina, che si basa sullo stesso concetto del labelling index con timidina tritiata, solo che l'evidenziazione non è condotta con autoradiografia poiché la bromodesossiuridina non è radioattiva, le cose vanno un po' meglio, ma c'è sempre il problema che occorre un tessuto vitale.
Per quanto riguarda l'indice mitotico, questa è una metodica sapientemente semplice, ma anche molto "beffarda". Infatti consiste nella semplice valutazione di un'area neoplastica con ingrandimento 400x su 10 campi continui; si conta il numero delle mitosi. E' chiaro che la valutazione è soggettiva; l'operatore può prendere per mitosi ciò che mitosi non è, e viceversa.
Un'ultima possibilità è rappresentata dal rilievo dei markers di proliferazione, che sono rappresentati fondamentalmente da PCNA e Ki67. Di che si tratta?
PCNA significa "Antigene Nucleare di Cellule Proliferanti"; è una proteina nucleare sintetizzata in fase S del ciclo cellulare, non espressa in G0, espressa anche da cellule non in ciclo (per accumulo precedente). Di solito un alto numero di PCNA è associato ad un alto numero di mitosi; la determinazione del PCNA può prescindere dall'uso di tessuto fresco, e non implica radioattività.
Ki67 è una proteina nucleare espressa in S, e associata alla DNA polimerasi. Se risulta presente in meno del 25% delle cellule, la prognosi è buona, mentre una sua presenza in più del 50% delle cellule depone per una condizione di malignità.
Rilevazione di acidi nucleici. Si possono rilevare acidi nucleici autoctoni o eterologhi (cioè di virus, batteri, funghi). E' possibile far questo in diversi metodi. Quale tecnica utilizzare? In realtà molto dipende da ciò che effettivamente si vuole fare dell'acido nucleico.
Se ci interessa ricercare la presenza/assenza di una determinata sequenza, e basta, allora la metodica da preferire è quella del "dot blotting"; se invece interessa effettuare un dosaggio quantitativo, allora si ricorre al "southern/northern blotting". Per amplificare la presenza di piccole quantità di acido nucleico, c'è la metodica della "PCR", mentre per localizzare la sequenza si ricorre alla "ibridazione in situ". Calma, andiamo avanti. Per capire un po' di che si tratta, dobbiamo far riferimento alle tecniche di ibridazione molecolare.
L'ibrido è un legame artificioso che si forma tra la sequenza da identificare e una sonda complementare, esterna, la cui composizione in sequenza è nota. Pertanto le tecniche di ibridazione molecolare presuppongono la esatta conoscenza della sequenza da studiare.
Panico! Traduciamo. Quello che ci interessa è trovare una determinata sequenza di acido nucleico. Noi sappiamo che gli acidi nucleici sono formati da sequenze di composti chimici chiamati nucleotidi: Adenina (A), Guanina (G), Timina (T), Citosina (C), Uracile (U). Sappiamo anche esistere la possibilità di un legame specifico tra A e T, e tra C e G. Quindi, una sequenza ad es., AATCTAG avrà come sequenza complementare TTAGATC. Giusto? Sì. Pertanto, se noi vogliamo individuare nel nostro campione la sequenza di acido nucleico AATCTAG, dobbiamo usare una sonda dalla sequenza TTAGATC, che vi si leghi. Starà a noi poi scegliere il modo per poter evidenziare l'avvenuto legame. Ora si può rileggere il paragrafo precedente. Va bene?
Le tecniche di ibridazione si dividono in non estrattive e estrattive. Esaminiamo adesso le tecniche non estrattive, o anche dette di ibridazione in situ. In queste tecniche la molecola di acido nucleico è svelata nel campione stesso, morfologicamente conservato.
In una prima fase si compie la permeabilizzazione delle membrane; questo serve per far entrare le sonde all'interno della cellula, dove ci sono gli acidi nucleici. Per ottenere il risultato, si usa un enzima proteolitico, che è la proteina kinasi k. A ciò segue una fase di denaturazione termica; questo, in realtà, vale solo per il DNA che, essendo una doppia elica, deve diventare a singola elica per permettere il legame della sonda. L'RNA, invece, è già monocatenario. Ad ogni modo, la denaturazione è condotta a 90-100 °C, sì da allontanare le eliche e far legare la sonda. La terza fase è di ibridizzazione, cioè si fa reagire la sonda marcata per un certo tempo a 37 °C. Segue quindi la procedura di stabilizzazione, che si effettua con dei "lavaggi di stringenza" per eliminare gli ibridi non legati o comunque non completamente legati. Questo punto del procedimento è molto importante, perché effettuando pochi lavaggi di stringenza si fa in modo che rimangano anche parecchie sonde non completamente legate (la metodica diventa poco specifica), mentre effettuandone molti rimangono solo le sonde completamente legate (la metodica si fa meno sensibile). Alla fine si effettua la evidenziazione del legame avvenuto con autoradiografia (se la sonda era radioattiva) o in microscopia ottica (se la sonda non era radioattiva ma altrimenti marcata).
Per marcare le sonde si possono utilizzare sostanza radioattive come 32P, 35S, 3H; si ha massima sensibilità, ma c'è rischio per l'operatore, inquinamento ambientale, veloce decadimento, tempi lunghi per l'autoradiografia. Le sostanze non radioattive, invece, come biotina, sulfoni, fluorocromi, sono più stabili e rapide, ma sono collegate ad una scarsa specificità, essendo rilevate in metodo indiretto. In ogni caso, le sonde devono essere complementari alla sequenza in studio e marcate. Sonde più grandi sono ovviamente più sensibili ma meno specifiche.
Per quanto riguarda invece le tecniche estrattive, in queste l'acido nucleico è estratto dal campione distruggendo lipidi e proteine con metodi chimici e/o enzimatici. Per ottenere questi risultati, occorre inizialmente omogeneizzare il campione, così da ottenere la distruzione delle membrane, dunque ottenere la precipitazione degli acidi nucleici dall'omogenato, e effettuare un dosaggio fluorimetrico della quantità di acidi nucleici. Quest'ultimo punto è importante perché ci dà informazioni sulla quantità di sonda da impiegare.
Ritornando a quelle che sono le caratteristiche delle sonde, facciamo qualche considerazione. Appare abbastanza logico che variando la lunghezza della sonda si può scegliere la specificità della metodica. Con sonde lunghe è possibile un legame con più sequenze. Facciamo un esempio. Supponiamo di voler riscontrare la presenza di un tipo particolare di HPV (Papilloma Virus Umano) in un tessuto. Esistono 74 diversi di questo virus. E' un virus epiteliotropo, che colonizza le cellule squamose vaginali, della cervice uterina e orali. A seconda del tipo di virus che ha infettato, ci possono essere lesioni diverse, però in alcuni casi si hanno quadri sovrapponibili. Ad es., HPV 6 e 11 danno condilomi intraepiteliali non invasivi, mentre HPV 16, 18, 31, 33 e 35 danno luogo a lesioni benigne che divengono aggressive.
In pratica. Una paziente "fa" il PAP test (quello citologico); se risulta "Betesda +"[13] e ha più di 45 anni, con tante gravidanze, si può togliere l'utero. Se invece la donna è più giovane, si va a ricercare HPV . Dapprima verranno utilizzate sonde più lunghe, per vedere se effettivamente c'è o non c'è HPV. Se la paziente è positiva per HPV, si utilizzano successivamente sonde più piccole per ricercare il tipo di HPV che ha determinato l'infezione. Poiché, è chiaro, ci sono sequenze piccole tipiche di un sol tipo di HPV e non di altri.
Dot blotting. Si tratta di una tecnica di ibridazione su filtro per DNA o RNA. E' una tecnica di ibridazione estrattiva. E' molto semplice, ma consente solo di valutare presenza o assenza. Come si procede? Innanzitutto si estrae l'acido nucleico e si posiziona il tutto in un pozzetto. Quindi si procede alla ibridazione con sonda, e successivamente a lavaggio. Quindi si evidenzia il legame avvenuto mediante tecnica di Southern blotting (per RNA) o Northern blotting (per DNA), che diferiscono per qualche dettaglio [non rivelato, NdR]. Più in dettaglio, una volta estratto l'acido nucleico, va sottoposto a reazione con enzimi di restrizione, che ne formano più frammenti a lunghezza prevedibile; quindi si procede ad elettroforesi sul gel di agarosio di tali frammenti, sì da separarli in base al p.m. Poi si compie il trasferimento dell'acido nucleico dal gel su un filtro di nitrocellulosa, nel quale rimanga intrappolato. Questo trasferimento si chiama "blotting". Qui va fatto reagire con una sonda marcata.
PCR. Reazione Polimerasica a Catena. Serve per amplificare una sequenza di acido nucleico. Estratto l'acido nucleico, viene tagliato con enzimi di restrizione. Quindi si aggiungono dei "primers", cioè piccole sequenze nucleotidiche, complementari ai punti estremi di tali sequenze. Si aggiunge poi basi, fosforo, zucchero e tampone adeguato, quindi DNA polimerasi. La DNA polimerasi potrà essere DNA o RNA dipendente. La RNA dipendenza è utilizzata quando si voglia amplificare una sequenza di RNA. La DNA polimerasi utilizzata è quella di Bacillus thermophilus, che vive in acqua calda e sulfurea a funziona fino a 70 °C. Questo particolare è importante. Dopo aver inserito nella macchina tutti questi reagenti, viene innescato un ciclo di reazioni di polimerizzazione dell'acido nucleico; ciclo che avviene ad elevata temperatura. Dopo una serie di cicli, si ottengono numerose copie dell'acido nucleico di partenza, anche se questo è in concentrazioni esigue.
Concetti generali di immunoistochimica
Tecniche
Quattro chiacchiere sull'Immunologia
Le applicazioni
Nozioni di biologia molecolare
[La microscopia elettronica, che non è in programma, probabilmente non verrà mai elaborata al computer]
[Forse uscirà una "II edizione" con aggiunte e correzioni secondo le lezioni di Bufo]
Con la microscopia elettronica si effettua l'indagine ultrastrutturale; questo consente di evidenziare direttamente la presenza di un virus, di definire quadri morbosi "nuovi", oncologici o meno, nel senso di una differenziazione ultrastrutturale non altrimenti indicabile, di riconoscere l'istogenesi delle neoplasie e di riconoscere l'etiopatogenesi di alterazioni non neoplastiche[15]. I settori di interesse della microscopia elettronica in Medicina sono parecchi: dermatologia, oncologia, nefrologia, anatomia patologica, neurologia, e così via.
Il microscopio elettronico ha una altissima risoluzione; per risoluzione si intende la distanza minima percepibile tra due punti distinti. Il potere di risoluzione per il microscopio ottico è di 0,2 , valore molto più grande rispetto al potere di risoluzione della microscopia elettronica.
Il microscopio elettronico quindi ha un elevato potere di risoluzione, un elevato potere di ingrandimento (fino a 4000 volte il valore dell'ingrandimento ottico tramite lente), di definizione (cioè di discernita nel contrasto tra punti chiari e punti scuri) e di penetrazione (cioè della capacità di attraversamento della luce attraverso il preparato).
In microscopia elettronica il prelievo, che ha dimensioni massime di 2-3 cm, viene valutato per un frammento che le dimensioni di 1 mm; si utilizza quindi un ultramicrotomo, che fornisce sezioni ultrafini (400-600 Angstrom[16]) e semifini (1000 Angstrom), che sono praticamente invisibili ad occhio nudo. Come mezzo per l'inclusione non si utilizza la paraffina, ma una durissima plastica.
Ma che si vede al microscopio elettronico? E' possibile condurre un esame dei singoli costituenti cellulari. Vediamo, vediamo, vediamo.
Endotelio. L'ultrastruttura dell'endotelio non è sempre la stessa. A livello epatico l'epitelio è discontinuo, e presenta pori; a livello renale l'endotelio è fenestrato, e presenta diaframmi; a livello nervoso, invece, è continuo (costituisce la barriera ematoencefalica!). In microscopia elettronica è possibile anche esplorare l'interstizio peri-endoteliale; ad es., nel fegato sarà visibile lo spazio di Disse, con tutti quei villi e microvilli aggettanti.
Cellule connettivali. E' tipica l'assenza delle giunzioni periferiche, che invece si riscontrano e sono ben visibili a livello delle cellule epiteliali.
Cromatina. Essa rappresenta il DNA, e può esistere come eucromatina (chiara, espressione di DNA rilassato, in cellule che non hanno tumulti proliferativi) o come eterocromatina (densa, espressione di DNA spiralizzato, in cellule che si stanno per moltiplicare; risulta addossata attorno alla membrana nucelare[17]).
Nucleoli. Hanno la funzione di sintetizzare rRNA. Sono organuli senza membrana, e risultano formati da regioni particolari dei cromosomi 13, 14, 15, 21 e 22, acrocentrici, che possiedono i geni del rRNA ripetuti più volte [sic]. I nucleoli contengono tre regioni: una zona fibrillare, una granulare, e un'altra, detta N.O.R. (Regione Organizzatrice del Nucleolo). In pratica i ribosomi vengono abbozzati nella zona fibrillare, e quasi completato nella zone granulare; quindi, nella zona granulare si forma RNA 45S [sic], nella zona granulare vi sono ribosomi più o meno maturi.
Membrana nucleare. In realtà è composta da due membrane distinte, che delimitano, come interspazio, la cisterna perinucleare. La membrana nucleare è in continuità con il reticolo endoplasmico, del quale è considerato come la prima parte; la membrana nucleare contiene dei pori che servono per la comunicazione nucleo-citoplasma.
Un reperto patologico relativo alla membrana nucleare è costituito dalla presenza di lamelle anulate, che nascono dalla sovrapposizione di più membrane nucleari; sono formazioni tipiche dei tumori secernenti.
Per inciso: quali sono le varie alterazioni ultrastrutturali delle cellule neoplastiche? E' possibile il riscontro di grossi nuclei, cromatina addossata alla membrana nucleare, grossi nucleoli, alterazioni della membrana nucleare (dovute al danno oncogeno a carico delle laminine); talvolta, è possibile riscontrare cumuli di glicogeno [sic].
Mitocondri. Gli elevati poteri di ingrandimento e risoluzione consente una visione ottimale di questi organuli al microscopio elettronico. I mitocondri si dividono contemporaneamente ed autonomamente rispetto alla divisione cellulare. I mitocondri, come si ricorderà, sono formati da due membrane: esterna e interna. La esterna è molto più permeabile di quella interna, risultando provvista di pori. La interna si ripiega a formare le creste mitocondriali, che a loro volta possono essere lamellari o vescicolari (quando, cioè, siano un po' dilatate). Nella faccia interna [non si sa di cosa, NdR] ci sono le ATPasi, formate da due subunità, F0, che sporge all'esterno, e F1, che invece sporge all'interno. Il tutto è ben visibile con il microscopio elettronico.
Sulla membrana mitocondriale interna vi sono inoltre numerosissimi altri complessi enzimatici; nei mitocondri vi sono inoltre depositi di calcio, ribosomi e DNA. Il DNA mitocondriale non possiede introni, ha un codice di lettura sui generis, e viene trasmesso secondo modalità diaginica; cioè il DNA mitocondriale che ognuno ha è quello dei mitocondri materni.
Sono presenti in gran numero soprattutto in quei siti dove occorre molta energia, ad es., nelle cellule dei tubuli renali. Nel muscolo si trovano di norma allineati in corrispondenza della banda "I"; nella sindrome di Duchenne, che è una malattia, si trovano invece vicino al nucleo.
Alterazioni patologiche osservabili per i mitocondri sono rappresentate da rigonfiamento, rimpicciolimento (specie nelle neoplasie), mancanza di creste o di componenti molecolari, inclusioni.
Perossisomi. Sono simili ai mitocondri, ma hanno solo una membrana, esterna; inoltre non hanno creste. Servono ad eliminare l'acqua ossigenata.
Ribosomi. Sono più piccoli rispetto ai granuli di glicogeno. Di solito sono aggregati nel citosol sotto forma di rosette, altre volte, invece, non sono aggregati tra loro, ma sono associati al reticolo endoplasmico. Aumentano molto come numero nelle cellule tumorali. Nel plasmocitoma, dove questo fatto è tipico, risultano più indifferenziati e la loro presenza correla con la capacità proliferativa.
RER. E' un insieme [sic] composto da vescicole, cisterne e tubuli. In realtà vi sono anche ribosomi, ma vi si attaccano un po' dopo la sua formazione. Il RER è coinvolto nella sintesi delle proteine; sintesi che può regolare grazie allo stato di un recettore, chiamato 7S. Reperti patologici involventi il RER sono dilatazioni e presenza di mitocondri all'interno.
REL. Anch'esso formato da vescicole, cisterne e tubuli, ma i ribosomi non vi si attaccano. Il REL serve alla sintesi di molecole steroidee, e infatti è presente soprattutto in quelle cellule che producono ormoni. Hanno inoltre anche funzione detossificante, poiché possiedono il citocromo P450.
Golgi. L'apparato di Golgi, presente in uno per cellula, si trova nelle vicinanze del nucleo, e serve alla maturazione delle proteine. Nella zona trans-Golgi ("al di là" del Golgi) vi sono vescicole più piccole, nella zona cis-Golgi ("al di qua") le vescicole preseneti sono più grandi.
Glicogeno. Può essere scambiato per ribosoma, ma il glicogeno è più grande. Il glicogeno può esistere in forma di aggregato (forma ) o meno (forma ).
Tutte le aggiunte ad immunoistichimica sono schematizzate a mano, e non sono presenti su questo file. Ciò contrariamente a quanto già scritto.
Non voglio perdere altro tempo.
Basta.
TUMORI DELLE PARTI MOLLI
introduzione
Per comune accezione, le parti molli del corpo umano comprendono i tessuti adiposo e fibroso, la muscolatura volontaria, i vasi e i nervi periferici; si tratta di strutture di derivazione prevalentemente mesodermica e solo in parte neuroectodermica. La più importante esclusione dalle parti molli - fra le componenti del connettivo - è il sistema linforeticolare, le cui neoplasie costituiscono un gruppo autonomo.
Pur potendo insorgere in qualsiasi area dell'organismo, tessuti di sostegno dei visceri compresi, le sedi principali di insorgenza dei tumori delle parti molli sono gli arti, i cingoli scapolare e pelvico, i tessuti sottotegumentari del tronco, del capo e del collo e la regione retroperitoneale.
I tumori benigni delle parti molli sono molto frequenti, ma hanno significato clinico limitato, essendo poco propensi all'infiltrazione e presentando lenta crescita espansiva con progressiva compressione dei tessuti circostanti.
I tumori maligni, detti anche sarcomi, sono meno frequenti di quelli benigni, secondo un rapporto inferiore a 1:100. Si può calcolare che in Italia si verifichino circa 1500-2000 nuovi casi di sarcoma l'anno. I sarcomi costituiscono meno dell'1% dei tumori maligni dell'età adulta; rappresentano invece quasi il 10% dei tumori maligni dell'età fino a 16 anni.
La bassa incidenza dei sarcomi contrasta con la molteplicità di tipi e sottotipi istologici, la maggior parte dei quali possiede significato clinico e prognostico proprio.
Fra i tumori benigni e i sarcomi trova posto un gruppo di tumori a bassa malignità potenziale che si distinguono istologicamente dalle forme benigne per un indice mitotico più elevato, ma inferiore a quello proprio dei sarcomi, e clinicamente per la crescita più rapida, la possibilità di recidive e, sia pur raramente, di metastatizzazione.
Di quest'ultimo gruppo fanno parte alcune fibromatosi dell'adulto e alcuni tumori vascolari (emangioendoteliomi ed emangiopericitomi).
Classificazione
I tumori benigni e maligni delle parti molli vengono classificati con un criterio istogenetico in base al tessuto adulto cui maggiormente assomigliano.
Pertanto, i principali tessuti e strutture di origine sono i seguenti: il tessuto fibroso (costituito da fibroblasti), gli istiociti tessutali in grado di assumere facoltativamente morfologia e funzioni di tipo fibroblastico, il tessuto adiposo, il tessuto muscolare striato e quello liscio, i vasi sanguigni e linfatici, i nervi e i paragangli.
Un'aliquota non trascurabile di sarcomi, tra cui il sarcoma epitelioide, il sarcoma alveolare, il sarcoma a cellule chiare e il sarcoma di Ewing, non è attribuibile attualmente ad alcun tessuto di origine.
Un altro gruppo, infine, presenta caratteri indistinguibili dai sarcomi solitamente a insorgenza dalle ossa e dalle cartilagini, come l'osteosarcoma e il condrosarcoma extrascheletrico.
|
Classificazione istogenetica dei tumori delle parti molli |
||
|
a) Tessuto fibroso b) Tessuto adiposo c) Istiociti d) Tessuto mm.striato e) Tessuto mm.liscio f) Vasi g) Nervi h) Paragangli |
Non attribuibili a nessun tessuto di origine |
A insorgenza da ossa e cartilagini |
|
i) Sarcoma epitelioide j) Sarcoma alveolare k) Sarcoma a cellule chiare l) Sarcoma di Ewing |
m) Osteosarcoma n) Condrosarcoma extrascheletrico |
|
Metodi di indagine diagnostica
Fermo restando che la diagnosi istopatologica di neoplasia delle parti molli viene posta nella maggior parte dei casi in base alle normali metodiche di colorazione (ematossilina, eosina), nel 10% dei casi la diagnosi può essere meglio precisata o addirittura resa possibile dal ricorso alla microscopia elettronica e alle metodiche immunoistologiche.
A livello ultrastrutturale, la dimostrazione per esempio di premelanosomi in un sarcoma a cellule chiare conferma la diagnosi; altrettanto dicasi della presenza di glicogeno nel sarcoma di Ewing, di linee Z nei rabdomiosarcomi e di granuli di neurosecrezione (oltre che di glicogeno) nel neuroblastoma e in alcune neoplasie neuroendocrine.
La principale limitazione all'impiego della microscopia elettronica è il campionamento forzatamente scarso e non sempre rappresentativo.
Tra i metodi immunoistochimici, possono essere d'aiuto quelli indicativi della natura endoteliale delle cellule di neoplasie vascolari (ricerca dell'antigene correlato con il fattore VIII) e della natura muscolare striata (ricerca della desmina e della mioglobina) nei rabdomiosarcomi formati da mioblasti non abbastanza differenziati per presentare la caratteristica striatura trasversale, e in genere tutti quelli che evidenziano le proteine dei filamenti intermedi e in particolare di vimentina.
I sarcomi sono quasi sempre neoplasie maligne fin dall'inizio e solo raramente derivano dalla trasformazione maligna di tumori connettivali benigni.
Un'eccezione a quest'ultima regola è la neurofibromatosi o malattia di von Recklinghausen, a trasmissione genetica autosomica dominante, in cui occasionalmente uno o più dei neurofibromi di cui il paziente è affetto si trasforma in schwannoma maligno.
Eccezionali sono invece le insorgenze di liposarcomi in lipomi, emangioendoteliomi in angiomi, fibrosarcomi in fibromi o fibromatosi. Deve essere inoltre precisato che la controparte benigna di alcuni sarcomi è di riscontro molto raro (es., rabdomiomi), mentre altri tumori benigni sono di comunissima osservazione (lipomi, leiomiomi, angiomi, istiocitomi fibrosi benigni).
Andamento delle neoplasie
I sarcomi sono caratterizzati da accrescimento progressivo con velocità variabile, in genere proporzionale al grado di malignità e di indifferenziazione, con graduale compressione delle strutture circostanti; queste vengono a formare una pseudocapsula intorno alla quale si costituisce un'area di tessuto di granulazione iperemico e ricco di infiltrati infiammatori.
La pseudocapsula consente spesso una facile enucleazione della neoplasia che al chirurgo non esperto dà la falsa impressione di benignità. In realtà, dopo pochi mesi, a distanza variabile dalla massa principale escissa e in misura proporzionale al grado di malignità del sarcoma, si formano noduli satelliti maligni, soprattutto in seguito alla migrazione di cellule neoplastiche lungo vasi e nervi o, eventualmente, lungo tragitti provocati da interventi chirurgici incompleti (ad es., biopsie con ago grosso).
Per tale motivo si ritiene attualmente che il metodo migliore di porre diagnosi istopatologica di sarcoma sia quello di effettuare un prelievo bioptico incisionale con successivo esame istologico del materiale incluso in paraffina.
In questo modo è solitamente garantita la rappresentatività del prelievo e la sufficienza quantitativa per eventuali altre indagini (microscopia elettronica).
|
SARCOMI |
|
|
Accrescimento |
Programmato a velocità variabile (in proporzione alla malignità) Presenza di una pseudocapsula Tessuto di granulazione iperemico e ricco di infiltrati infiammatori |
|
Velocità di disseminazione |
Dimensioni Profondità della sede Grading Terapia errata |
|
Ripresa della malattia |
47% recidiva locale 38% metastasi a polmone 9% metastasi a linfonodi, cute, ossa |
|
Metastasi |
Facilmente per via ematica Per via linfatica s.sinoviale, rabdomiosarcoma sinoviale, s. a cellule chiare, s. epitelioide, angiosarcoma |
Sono invece da proscrivere i prelievi escissionali e non sono raccomandabili sia i prelievi con ago sottile (diametro inferiore a 1 mm) per esame citologico su striscio, sia quelli con ago grosso (diametro superiore a 1 mm) per la scarsa rappresentatività del materiale ottenuto in rapporto al frequente polimorfismo di molti sarcomi.
I prelievi con ago trovano invece indicazione più precisa per il monitoraggio di pazienti con malattia nota e per la diagnosi di eventuale ripresa di malattia in sede di intervento (recidiva) o a distanza (metastasi).
L'esame preoperatorio del prelievo bioptico, al microtomo criostato o congelatore, è utile in mani esperte, tuttavia non è indispensabile, potendo l'intervento definitivo attendere il normale esame istologico.
La velocità di disseminazione dei sarcomi è in genere proporzionale alle dimensioni, alla profondità della sede (sopra o sotto fasciale) e al grado di differenziazione istologica. Altri fattori che influiscono sono gli eventuali interventi terapeutici erronei, la risposta biologica dell'ospite e l'età del paziente; per es., fibrosarcomi morfologicamente simili manifestano maggiore aggressività nell'adulto che nel bambino.
Tra le forme di ripresa di malattia, la recidiva locale è la più frequente (47%), seguita dalle metastasi al polmone (38%), ai linfonodi (9%), alla cute e alle ossa.
La disseminazione a distanza (metastatizzazione) avviene prevalentemente per via ematica, con destino preferenziale per i polmoni, il fegato e le ossa, in ordine decrescente di frequenza.
La disseminazione per via linfatica non è rara, è per lo più tardiva e riguarda solo alcuni tipi di sarcomi. I sarcomi maggiormente linfotropi sono il sarcoma sinoviale, il rabdomiosarcoma alveolare - che raggiunge i linfonodi regionali entro il primo anno di malattia nel 75% dei casi -, il sarcoma a cellule chiare, il sarcoma epitelioide e l'angiosarcoma (emangioendotelioma maligno).
La tendenza a produrre metastasi linfonodali è comunque sempre largamente inferiore a quella dei carcinomi e dei melanomi maligni.
Stadiazione
Ai fini di una corretta valutazione dell'estensione della malattia (stadiazione) e di un trattamento chirurgico adeguato, è stato introdotto da Enneking il concetto di compartimento anatomico, che corrisponde a una struttura ben definita, quale un'articolazione o una loggia muscolare delimitata da fasce. I sarcomi a insorgenza intracompartimentale arrivano a oltrepassare i limiti del compartimento solo tardivamente, mentre quelli extracompartimentali, meno frequenti, si diffondono più precocemente; altrettanto rapida è la diffusione quando un sarcoma da intra è diventato extra compartimentale.
Il significato terapeutico del compartimento è importante poiché soltanto la resezione compartimentale è da considerare radicale, mentre la resezione i cui margini siano al di fuori della pseudocapsula, ma cadano all'interno del compartimento, viene definita ampia. E' marginale la resezione coincidente con la pseudocapsula e intralesionale quella condotta nel contesto del tessuto neoplastico.
Le probabilità di recidive locali sono tanto minori quanto più esteso è l'intervento, e poiché la ripresa di malattia è caratterizzata dall'accrescimento dei noduli satelliti sopradescritti, ne deriva che l'aspetto clinico e macroscopico caratteristico delle recidive di sarcoma a seguito di interventi inappropriati è costituito da una nodularità multipla sotto e paracicatriziale insorgente dopo circa tre mesi dall'intervento non radicale.
Il grado di malignità istologica di un sarcoma è uno dei maggiori fattori prognostici insieme alla sede superficiale o profonda e alle dimensioni, maggiori o minori di 5 cm di diametro.
Nella valutazione del grado di malignità istologica (grading) si tiene conto della densità cellulare, dell'estensione dei fenomeni necrotici, della maturazione delle cellule nel senso di una maggiore o minore somiglianza alle cellule del tessuto normale corrispondente e, soprattutto, dell'indice mitotico.
In genere i tumori benigni presentano 1-2 cariocinesi per 10 campi microscopici a 400 ingrandimenti, mentre i tumori maligni ne presentano da 5 in su con punte di 20-30. Occorre precisare al riguardo che il conteggio delle mitosi, sia atipiche che normalmente configurate, riflette la realtà solo se il tessuto escisso è stato fissato immediatamente. In caso di fissazione ritardata, il ciclo mitotico si completa e l'indice scende a valori prossimi allo zero.
Il grading, solitamente suddiviso in tre categorie, è applicabile utilmente ad alcuni sarcomi, quali il leiomiosarcoma, l'emangiopericitoma e il fibrosarcoma, mentre la maggior parte dei sarcomi presenta un proprio grado istologico di malignità legato all'istotipo.
Sono sempre a basso grado di malignità il fibrosarcoma dell'infanzia, il dermatofibrosarcoma protuberans, i liposarcomi differenziato e mixoide e l'emangioendotelioma epitelioide.
Sono sempre ad alto grado di malignità i liposarcomi a cellule rotonde e polimorfo, l'istiocitoma fibroso maligno (a eccezione della varietà mixoide che è di grado intermedio), il rabdomiosarcoma, indipendentemente dalla varietà istologica, alcuni angiosarcomi, il sarcoma sinoviale, lo schwannoma maligno, il condrosarcoma mesenchimale, l'osteosarcoma e il sarcoma di Ewing extrascheletrici.
Presentano grado intermedio di malignità l'istiocitoma fibroso maligno mixoide, il sarcoma di Kaposi, nella sua varietà tradizionale, alcuni angiosarcomi, il condrosarcoma mixoide, il tumore a cellule granulari maligno (cosiddetto mioblastoma), il sarcoma alveolare delle parti molli, il sarcoma epitelioide e il sarcoma a cellule chiare.
Altre notizie
Per quanto riguarda la classificazione istopatologica dettagliata, si rimanda alle trattazioni specialistiche. Gli Autori che maggiormente si sono occupati dell'argomento sono Stout negli anni '50, Lattes negli anni '60 e Enzinger negli anni '70. Con l'apporto decisivo di questi ultimi due studiosi è stata elaborata la classificazione istopatologica internazionale sotto l'egida dell'OMS e pubblicata nel 1969.
Da allora, tuttavia, si sono verificati alcuni mutamenti nei criteri diagnostici e classificativi soprattutto con la creazione della categoria dei tumori fibroistiocitici, istogeneticamente non lontani dai tumori del tessuto fibroso. Le modificazioni dei criteri diagnostici avvenute col tempo hanno fatto sì che le frequenze relative dei vari tipi istologici siano cambiate negli ultimi decenni, non consentendo facili confronti rispetto alle eventuali reali modificazioni di incidenza.
A titolo di esempio, valga l'incidenza del 30% del fibrosarcoma, de11' 8% del rabdomiosarcoma e del 3% del sarcoma sinoviale nel 1950, contro quella del 10% del fibrosarcoma, del l5% del rabdomiosarcoma, quasi del 20% dell'istiocitoma fibroso maligno e del 10% del sarcoma sinoviale negli anni '70.
Un'entità un tempo frequentemente diagnosticata, il mixoma e le sue varie combinazioni (fibromixoma, mixolipoma), è attualmente quasi scomparsa dalle classificazioni, a eccezione di alcuni mixomi intramuscolari, della mandibola, del mascellare e delle guaine nervose, essendo stata riconosciuta la frequentissima presenza nello stroma di una componente mixoide di molte neoplasie, quali il liposarcoma (liposarcoma mixoide), il condrosarcoma (condrosarcoma mixoide) e l'istiocitoma fibroso maligno (varietà mixoide) e la presenza occasionale di stroma mixoide in altri sarcomi, quali il leiomiosarcoma e lo schwannoma maligno.
Nell'ambito delle neoformazioni del tessuto fibroso, oltre ai tumori benigni e maligni, esistono lesioni simil-tumorali che non di rado causano errori diagnostici clinici e istopatologici.
Si tratta di lesioni nodulari a rapida crescita, di diametro solitamente inferiore a 3 cm, spesso modicamente dolenti, per lo più solitarie, ubiquitarie, più frequenti in giovani adulti, e che solo in una parte dei casi sono precedute da traumi.
Macroscopicamente la somiglianza con una neoplasia è data dall'aspetto nodulare non capsulato, anche se ben delimitato, che ha fatto coniare il termine di fascite nodulare, talora definita anche pseudosarcomatosa, proliferativa o infiltrativa.
Le sedi di insorgenza più frequenti sono il tessuto sottocutaneo, la muscolatura volontaria e le fasce superficiali, soprattutto delle estremità superiori e inferiori nell'adulto e del capo e del collo nei bambini.
Le difficoltà diagnostiche istopatologiche sono legate alla presenza di una cellularità fibroblastica vivacemente proliferante e immersa in uno stroma mixoide o fibroso, che spesso dissocia, infiltrandoli, i tessuti circostanti, muscolatura striata compresa.
Lesioni analoghe sono la miosite proliferativa, caratteristica dei ventri dei muscoli volontari e, verosimilmente, anche il cosiddetto (angio)fibroma delle guaine tendinee o tenosinoviale delle mani e dei piedi di soggetti di sesso maschile e che è oggi considerato un esito tardivo di fascite nodulare. Si tratta di noduli piccoli e circoscritti e talora dolenti, costituiti da fibre collagene ammassate nel centro e da fibroblasti e miofibroblasti alla periferia. Anche l'assenza di recidive dopo asportazione completa dimostra la natura non neoplastica di questo gruppo di lesioni.
NEOFORMAZIONI DEL TESSUTO fibroso
Distinguiamo forme benigne, forme maligne, e poi delle lesioni similtumorali. Trattiamo qui di quest'ultime.
Le lesioni similtumorali sono lesioni nodulari a rapida crescita, dal diametro inferiore ai 3 cm, solitarie, dolenti; hanno possibilità di localizzazione ubiquitaria, e colpiscono principalmente giovani e adulti. Una volta asportate, non danno luogo a recidiva.
Si presentano ad aspetto nodulare non capsulato, ben delimitato ("fascia nodulare"), in sede sottocutanea, o a livello della muscolatura volontaria, o sulle fasce superficiali (nei bambini frequente la localizzazione al collo). Microscopicamente si osserva una cellularità fibroblastica ad alta proliferazione, immersa in stroma mixoide; la neoformazione riesce a dissociare i tessuti circostanti.
Queste lesioni similtumorali, comunque, non riguardano soltanto il tessuto fibroso; ad esempio, si ritrovano come emangioendotelioma intravascolare vegetante di Masson in emorroidi e polipi laringei.
Classificazione
I tumori fibrosi contemplano forme benigne, le fibromatosi e il fibrosarcoma.
Tra i tumori benigni troviamo il fibroma, il cheloide, l'elastofibroma e il fibroblastoma a cellule giganti. Le fibromatosi, invece, si dividono in due grandi categorie: le forme dell'adulto e quelle infantili, nelle ambito delle quali si individuano numerosi sottogruppi. Le fibromatosi dell'adulto possono essere superficiali o profonde. Le forme superficiali contemplano la fibromatosi di Dupuytren, della pianta del piede e del pene; le forme profonde, invece, si dividono in addominali ed extra-addominali. Le fibromatosi infantili, invece, non hanno una suddivisione così schematica. Il fibrosarcoma è una forma maligna.
Forme benigne
Il fibroma è una neoformazione dermica, dal diametro che arriva al cm; il più delle volte presenta un peduncolo. Esiste un fibroma duro e un fibroma molle, a seconda della composizione in fibre collagene (rispettivamente bassa o elevata). I fibromi possono presentarsi nell'ambito di sindromi definite, come, ad es., la sindrome di Gardner, con poliposi intestinale, osteomi, cisti cutanee.
Il cheloide è un ammasso di fibre collagene variamente intrecciate, situato nel derma e con tendenza alla calcificazione e ossificazione. E' l'esito di una cicatrizzazione esuberante.
L'elastofibroma risulta composta invece da fibre collagene e elastiche degenerate; ha sede nei tessuti molli profondi, particolarmente sul dorso, e colpisce soprattutto soggetti anziani.
Il fibroblastoma a cellule giganti è composto da fibroblasti e cellule giganti fusate bizzarre e polimorfe, in abbondante stroma mixoide. Si localizza nel sottocutaneo, in soggetti giovani; tende a recidivare.
Fibromatosi dell'adulto
Le fibromatosi sono un gruppo di lesioni non classificabili per caratteristiche biologiche come tumori benigni, ma nemmeno come neoplasie maligne.
Le fibromatosi dell'adulto superficiali non hanno alcuna tendenza alla trasformazione maligna; in alcuni casi si ravvisano delle somiglianze istologiche con i fibrosarcomi. Hanno rapida crescita, seguita da una fase cicatriziale retraente. Alcune forme hanno carattere familiare, e hanno localizzazione bilaterale (è il caso delle forme plantari e palmari).
Nella malattia di Dupuytren, maggiormente diffusa presso i bianchi maschi, si realizza una associazione con noduli fibrosi sulle articolazioni interfalangee prossimali e sulle articolazioni metacarpofalangee.
Le fibromatosi dell'adulto profonde si distinguono in una forma addominale e una forma extra-addominale.
La forma addominale riguarda maggiormente le donne, dopo una gravidanza. La lesione fibromatosa ha sede a livello delle strutture muscolo-aponeurotiche dei muscoli retto e obliquo interno. Si tratta di un fenomeno neoplastico localmente invasivo, che recidiva nel 20-20% dei casi, ad etiologia traumatica in soggetti predisposti.
Più in particolare, sono anche descritte delle fibromatosi pelviche e mesenteriche, che danno segno di sé simulando un dolore rispettivamente ovarico o intraddominale.
La forma extra-addomianale, che è tra l'altro la forma più aggressiva (da un punto di vista biologico) tra le fibromatosi, colpisce soggetti in età giovane, a spalla, parete toracica, dorso, coscia, ginocchio, collo. Istologicamente si distingue una massa neoplastica delimitata dalle dimensioni cospicue, con fibroblasti non particolarmente atipici e abbondante collageno disposto a fasci ondulati. Nel 50% dei casi si realizza una recidiva. L'etiologia prevede una precedente esposizione a traumatismo chirurgico o a irradiazione (comunque in sede di ferite chirurgiche).
Fibromatosi infantili
Descriveremo 8 quadri diversi di fibromatosi.
Amartoma fibroso dell'infanzia - Colpisce bambini molto piccoli (1 anno), in sede ascellare. Istologicamente si nota un tessuto fibroso misto a lobuli di tessuto adiposo maturo e immaturo; il tutto è mal delimitato.
Fibromatosi digitale infantile - Colpisce bambini a 2 anni di età, alle dita delle mani e dei piedi; può essere multipla, bilaterale, può recidivare subito o regredire spontaneamente. Istologicamente si osserva proliferazione di fibroblasti e miofibroblasti con inclusioni citoplasmatiche eosinofile simili ad eritrociti; sono costituite da filamenti di actina.
Miofibromatosi - Colpisce poco dopo la nascita. Esiste una variante solitaria, più frequente, a derma, sottocutaneo, muscoli, e una forma multicentrica, in cui compaiono miofibromi anche in visceri e ossa. Si tratta di noduli dal diametro al max. di 2 cm, in cui vi sono fasci di fibro e miofibroblasti, insieme a focolai di necrosi e calcificazione. Sono formazioni autolimitanti.
Fibromatosi ialina giovanile - A carattere ereditario, simile alla malattia di von Recklinghausen, è causa di deformità del capo; può essere associata a ritardo mentale.
Fibromatosi del collo - Colpisce nelle prime settimane di vita, e la sede è il terzo inferiore del muscolo sternocleidomastoideo. Ha una rapida crescita iniziale, che poi rallenta fino a regredire (!). Dopo la escissione non vi sono recidive. Forse è causato dal parto traumatico. Istologicamente si realizza una proliferazione sierosa che infiltra e sostituisce le fibre muscolari.
Fibromatosi infantile desmoide - Colpisce nei primi due anni di vita. La sede può essere addominale, o alla lingue, o alla parte alta del collo; si realizzano deformità articolari e ossee. Tende a recidivare, e non regredisce spontaneamente. Dal punto di vista istologico, vi sono cellule con caratteri di diversa maturità.
Fibroma aponeurotico (calcificante) - Localizzato a mani e piedi, è una lesione unica, che tende a recidivare, ma non si trasforma in tumore maligno. Il diametro di tale lesione, nodulare, è di qualche cm; è composto da tessuto fibroso compatto con zone cartilaginee e calcifiche.
Fibromatosi gengivale - Anche detta ipertrofia gengivale, è una caratteristica ereditaria (autosomica dominante), in cui si realizza una lenta crescita di tumefazioni costituite da tessuto fibroso simile a una cicatrice. La sede è gengivale e al palato duro. Non inficia negativamente la eruzione dei denti.
Fibrosarcoma
Un tempo molto più frequente che ora; questo era dovuto al fatto che nella menzione di fibrosarcoma erano contemplate delle forme che oggi sono nettamente separate (schwannoma maligno, sarcoma sinoviale monofasico, istiocitoma fibroso maligno).
Colpisce intorno ai 30-60 anni, a coscia, ginocchio; meno frequentemente a tronco, braccia, gambe. Può comparire in seguito a terapie radianti. Presenta una struttura a fasci intrecciati di cellule fusiformi, con disposizione a spina di pesce; vi è collagene. Il numero delle mitosi è in dipendenza dalla malignità. Si osservano ancora calcificazioni metaplastiche, fenomeni necrotici, emorragici; dà luogo a metastasi ematogene a polmoni e scheletro (vertebre, cranio). E' possibile differenziare il fibrosarcoma dal sarcoma sinoviale per la assenza di cheratina nelle cellule, e dallo schwannoma maligno per l'assenza della proteina S100 nelle cellule di Schwann.
Esiste anche un fibrosarcoma infantile. Si osserva dalla nascita o durante il primo anno di vita, principalmente nei maschi, e preferibilmente alle parti distali degli arti. Dalle dimensioni variabili, il suo grading non è mai superiore a 2 (II° grado), e il collagene è quasi sempre sotto forma di fibrille reticolari.
TUMORI FIBROISTIOCITICI
Originano da istiociti che assumono caratteristiche morfologiche e funzionali di fibroblasti.
Classificazione
Si distinguono delle forme a basso grado di malignità e le varianti dell'istiocitoma fibroso maligno.
Tra le forme a basso grado di malignità vi sono l'istiocitoma fibroso benigno della cute, l'istiocitoma fibroso profondo (IFP), lo xantogranuloma giovanile, il reticoloistiocitoma nodulare, gli xantomi, il dermatofibrosarcoma protuberans, il fibroxantoma atipico.
L'istiocitoma fibroso maligno, invece, prevede le varietà storiforme/polimorfa, mixoide, a cellule giganti, infiammatoria, angiomatoide.
Forme benigne
L'istiocitoma fibroso benigno della cute ha un diametro che non supera i 3 cm., è spesso una lesione multipla, e localizzata alle estremità, nel derma o nel sottocutaneo.
L'istiocitoma fibroso profondo (IFP) ha un diametro non superiore ai 5 cm; la diagnosi si basa sulla assenza di mitosi e atipie; difatti osservata sul vetrino la lesione presenta margini mal delimitati, e fasci fibrosi intrecciati. La cellularità è composta da fibroblasti, istiociti con citoplasma schiumoso (in attività fagica), da cellule giganti; i vasi dimostrano una parete ialinizzata. In realtà tutti i caratteri finora descritti sono presenti in tutti gli istiocitomi. Ciò che caratterizza l'IFP è la struttura storiforme (a vortice).
Lo xantogranuloma giovanile è multiplo; compare alla nascita come papule rossastre localizzate a capo, collo, estremità. Sono presenti istiociti, cellule giganti, eosinofili. Forse si tratta di una lesione reattiva ad agenti peraltro ignoti. E' anche chiamato nevoxantogranuloma, poiché è simile ad un nevo.
Il reticoloistiocitoma nodulare dell'adulto può esistere in forma nodulare (in tal caso benigna e autolimitantesi) o in forma pluricentrica (associata a sintomatologia sistemica e artrite). Si vedono, al microscopio, istiociti grossi e plurinucleati, il cui citoplasma è ricco in lipidi e glicoproteine.
Gli xantomi sono raccolte circoscritte di istiociti contenenti lipidi nel citoplasma. Clinicamente si distinguono forme eruttiva, tuberose, piane, tendinei, e poi ci stanno anche gli xantelasmi. Istologicamente, però, sono la stessa cosa.
Il dermatofibrosarcoma protuberans ha sede cutanea o sottocutanea, al tronco e alla parte prossimale delle estremità. Colpisce in età adulta, ed ha una crescita lenta, progressiva, infiltrante. Si tratta di noduli che retraggono sino ad ulcerare l'epidermide. Non presentano una capsula. Vi sono fibroblasti disposti a vortice, rare cellule giganti, abbondante sostanza fondamentale con mucine acide (non sempre). Quando si trova una pigmentazione melanica intracitoplasmatica, si parla di tumore di Bednar.
Il fibroxantoma atipico, a decorso lento, è istologicamente indistinguibile dall'istiocitoma fibroso maligno. Ha sede a cute di capo e collo, tronco e arti di soggetti giovani.
Istiocitoma fibroso maligno
Colpisce principalmente soggetti di sesso maschile, con più di 50 anni.
Ha sede a livello della muscolatura scheletrica sottofasciale delle estremità, e anche nello spazio retroperitoneale.
Macroscopicamente esso si presenta come una massa plurilobata dal diametro superiore ai 5 cm; questa formazione è fornita inizialmente di pseudocapsula, ma con l'andar del tempo diviene infiltrante. Ha un aspetto gelatinoso-compatto, e un colorito che varia dal giallastro al marezzato grigio-emorragico.
Da un punto di vista della microscopia, possiamo individuare 5 varianti:
Storiforme - polimorfo. Presenta cellule simili a fibroblasti senza eccessive atipie, disposte a vortice, e cellule simili ad istiociti, voluminose, con nuclei atipici. Nel citoplasma si riscontrano granuli PAS+, diastasi-resistenti, e grassi neutri. C'è inoltre assenza della proteina S-100, e positività per lisozima, -1AT, 1ACT e vimentina. Lo stroma presenta fibre collagene ialinizzate, e sottili fibrille reticolari. Metastasi vengono inviate a polmoni, linfonodi, fegato, ossa. Il grado di malignità dipende dalle dimensioni del tumore e dalla sede (particolarmente pericolose sono le sedi profonde e/o più vicine al centro del corpo).
Mixoide. In questa varietà si riscontra ricchezza di sostanza fondamentale, ricco in mucopolisaccaridi acidi; si tratta di una sostanza sensibile alla digestione con ialuronidasi.
Infiammatoria. Detto anche xantogranuloma, ha sede retroperitoneale. Istologicamente si osservano istiociti con citoplasma schiumoso (lipidi), cellule giganti plurinucleate, fibroblasti, numerosi neutrofili, linfociti, plasmacellule.
Tumore gigantocellulare delle parti molli. Si caratterizza per la presenza di elementi cellulari giganti.
Angiomatoide. Forma rara, appare spesso in soggetti giovani. Simula un ematoma o un vaso trombizzato, si presenta con un aspetto multinodulare. Al microscopio si osservano cellule istiocitiche, cellule flogistiche (cellule della flogosi cronica) e marcati fenomeni emorragici, con formazione di spazi cistici rivestiti di istiociti, e non da endotelio.
tumori lipomatosi
Anche in questo caso distingueremo forme benigne e forme maligne; le forme maligne sono rappresentate dal liposarcoma nelle sue varietà.
Forme benigne
Lipomi. Costituiscono l'80% delle neoplasie adipose benigne, e sono piuttosto caratteristiche della donna in età adulta (sedi preferenziali dorso, spalla, collo, cingoli). Microscopicamente si realizza la presenza di adipociti maturi, di una componente vascolare ben rappresentata; da notare la assenza di lipoblasti.
Angiolipoma. Particolarmente frequente all'avambraccio e dolorabile quando palpato, presenta una ricca componente vascolare in tessuto lipomatoso.
Lipoblastoma benigno. Tipico dell'età infantile, si presenta frequentemente nelle estremità. Si tratta di una neoformazione solitaria. Quando, però, le formazioni sono diffuse e infiltrano la muscolatura volontaria, si parla meglio di lipoblastomatosi.
Lipoma intramuscolare. Colpisce principalmente maschi adulti, e risulta composto da adipociti senza atipie che dissociano la muscolatura striata.
Liposarcoma
La massima incidenza si riscontra in età medio-avanzata (dal 5° al 7° decennio di vita), in sede rappresentata da estremità inferiori e spazio retroperitoneale. Si tratta di masse molto voluminose che microscopicamente possono presentare quadri diversi.
Liposarcoma ben differenziato. Presenta lipoblasti e zone ricche in elementi flogistici; questo liposarcoma può recidivare.
Mixoide. E' la forma più frequente, e presenta un'elevata quota di matrice mixoide, lipoblasti a diverso stadio di maturazione, e ampi capillari variamente ramificati.
Liposarcoma a cellule rotonde. Presenta cellule rotonde con poco citoplasma vacuolizzato; può essere considerato come una forma meno differenziata rispetto alla forma mixoide, di cui sopra. Questa forma dà luogo a metastasi per via ematogena.
Liposarcoma polimorfo. Anch'esso metastatizzante. Si riconosce la presenza di lipoblasti maligni. Queste cellule sono caratterizzate dal fatto di essere tutte uguali fra loro; non accumulano lipidi citoplasmatici, ma presentano vacuoli otticamente vuoti. Inoltre vi è positività alla apoproteina S100, e vi sono filamenti di vimentina nel citosol.
tumori della muscolatura liscia
Le forme benigne sono rappresentate dai leiomiomi, le forme maligne dai leiomiosarcomi.
Leiomiomi
Distinguiamo 4 categorie diverse di leiomiomi: cutanei, solitari, vascolari, epitelioidi.
Cutanei. I leiomiomi cutanei riguardano i muscoli erettori dei peli e hanno sede dermica. Sono dolorabili, multipli, e localizzati più spesso alle estremità, ai genitali esterni, alle areola mammarie, ai capezzoli.
Solitari. Localizzati alle aree genitali maschili e femminili, provocano dolore spontaneo.
Vascolari. Anche detti angiomiomi, sono formazioni solitarie, sottocutanee, dolenti. Caratterizzati da proliferazione di cellule muscolari lisce con vasi a parete simile alle arteriole, ma privi di lamine limitanti elastiche.
Leiomiosarcoma
Ha sede retroperitoneale o in cavità addominale (in questi due casi la localizzazione è dunque profonda), ma sono descritte anche forme localizzate a derma e sottocute (in tal caso la localizzazione è superficiale). Colpisce soprattutto donne con più di 60 anni.
Si tratta di neoformazioni voluminose, passibili di infezione, tendenti alla recidiva (più del 50% dei casi), e con probabilità di metastasi. A questo proposito, c'è da dire che le forme che più frequentemente metastatizzano sono quelle profonde (40%), mentre più raramente quelle superficiali (cutanee, 10%; sottocutanee, 10%).
Il leiomiosarcoma è formato da cellule allungate, con abbondante citoplasma eosinofilo, e nucleo centrale con estremità tozze. La presenza di modeste atipie nucleari conferisce il "grado I°" di differenziazione, mentre nuclei ipercromatici, abnormi e bizzarri fan parlare di "grado II°". Il citoplasma presenta delle striature, espressione della presenza di filamenti; vi è espressione contemporanea di desmina e vimentina.
I leiomiosarcomi epitelioidi (o leiomioblastomi maligni) si presentano microscopicamente con una predominanza della commistione di cellule epitelioidi e cellule fusate; caratteristica l'assenza di miosina.
Vi sono poi i leiomiosarcomi della parete dei vasi; particolarmente nelle vene (cava inferiore, safena, iliaca, femorale), meno frequentemente nelle arterie (polmonare).
tumori della muscolatura striata
Le forme benigne sono rappresentate dai rabdomiomi, quelle maligne dai rabdomiosarcomi. In ogni caso, è possibile distinguere dei sottotipi.
Rabdomiomi
I rabdomiomi sono tumori benigni della muscolatura striata. Si distinguono essenzialmente tre tipi:
a) Tipo adulto; a sede in capo e collo, negli anziani.
b) Tipo fetale; la sede è come nel tipo adulto, ma si realizza nell'età infantile.
c) Del cuore; in realtà è più un amartoma che una neoplasia; si tratta di una condizione associata alla sclerosi tuberosa.
Rabdomiosarcomi
Il rabdomiosarcoma è un tumore non epiteliale abbastanza frequente nelle prime due decadi di vita.
Anche i rabdomiosarcomi sono divisi in diverse tipologie, stavolta però con una differenza in base a criteri di microscopia (embrionale, alveolare, polimorfo).
Sebbene vi siano delle sedi effettivamente frequenti, sono descritte anche sedi un po' più particolari, come la regione testicolare (possibile invasione dei linfonodi retroperitoneali), l'orbita (causa uno spostamento del bulbo oculare, con invasione delle palpebre e parti ossee), la vescica urinaria.
Macroscopicamente appaiono mal delimitati, e spesso plurinodulari, polipoidi. Hanno un aspetto gelatinoso grigio-biancastro. Nel loro contesto possono verificarsi fenomeni emorragici, necrotici, cistici.
Hanno caratteristicamente un alto grado di malignità, e danno recidive per completa asportazione; possono dar luogo a metastasi a polmone e linfonodi. Anzi, le metastasi ai linfonodi sono frequenti e anche precoci.
Rabdomiosarcoma embrionale. Costituisce il 60% dei fenomeni, è frequente in soggetti con meno di 15 anni, a capo, collo, apparato genitourinario, retroperitoneo, alle estremità. Presenta delle aree dense, ricche in cellule, e delle aree meno dense, dove c'è un fondo mixoide. Le cellule hanno un aspetto ellissoidale o fusato, hanno scarso citoplasma e un nucleo ipercromatico. I nucleoli sono piccoli nelle cellule che appaiono meno differenziate. Le cellule che qui si trovano, rabdomioblastiche, possono essere anche voluminose con un citoplasma molto eosinofilo. I rabdomioblasti, poco maturi, hanno positività per la desmina; cellule più mature, invece, hanno positività per miosina e mioglobina. Il sarcoma, quando può espandersi senza costrizione del tessuto circostante assume aspetto polipoide, e viene anche chiamato sarcoma botrioide.
Alveolare. Rappresenta il 30% dei rabdomiosarcomi. Sede a capo, collo, apparato genitourinario, retroperitoneo, alle estremità, quindi similmente alla forma embrionale. Da un punto di vista microscopico presenta un connettivo fibroso denso, che delimita spazi rivestiti da rabdomioblasti; nel centro delle strutture, alveolari, vi sono cellule patologiche degenerate. Rare le cellule mature. I nuclei appaiono lobulati e ipercromatici. Vi è anche frequentemente presenza di cellule plurinucleate. Interessante il dettaglio per cui nelle forme metastatiche viene comunque mantenuta la struttura a spazi alveolari.
Rabdomiosarcoma polimorfo. Si riscontra in soggetti con più di 50 anni, e molto frequentemente alle estremità. In questa forma sono rare le cellule con striatura trasversale, mentre invece quello che è frequente è il riscontro di cellule con glicogeno citoplasmatico; accanto a queste, possono rimanere inglobate cellule muscolari striate che sono perfettamente normali.
tumori vascolari
Classificazione
Oltre a forme benigne e a forme maligne, i tumori dei vasi sanguiferi contemplano anche forme particolari, "di mezzo", gli emangioendoteliomi, che sono dei tumori vascolari a bassa malignità potenziale. I tumori dei vasi linfatici, invece, vanno sotto il nome generico di linfangiomi.
I tumori vascolari benigni (sia di vasi sanguiferi che di vasi linfatici - i linfangiomi -) possono essere circoscritti o diffusi. Le forme diffuse vanno sotto il nome di angiomatosi (per i vasi sanguiferi) e linfangiomatosi (per i vasi linfatici), mentre le forme circoscritte possono presentare diverse morfologie.
I tumori vascolari maligni, invece, contemplano gli angiosarcomi (o emangioendoteliomi maligni), il sarcoma di Kaposi, il tumore glomico, gli emangiopericitomi.
Emangiomi
La sede più frequente degli emangiomi è la cute, mentre meno frequentemente ci capiterà di osservare un emangioma in sede epatica, o cardiaca, o ossea, oppure al SNC. L'emangioma può essere capillare, cavernoso, racemoso.
Capillare. Si tratta di formazioni multiple e pianeggianti che col tempo divengono rilevate e infine spariscono fisiologicamente. Al microscopio si osservano spazi vascolari sottili rivestiti da endotelio normale ma voluminoso. Una sua variante è il granuloma piogenico, tendente alla ulcerazione e alle sovrainfezioni che lo rendono simile al tessuto di granulazione teleangectasico.
Cavernoso. Formazione voluminosa, se lesionata può provocare emorragie anche fatali. Microscopicamente si osservano ampi spazi vascolari di tipo venoso con parete sottile, insieme a fenomeni trombotici.
Racemoso. Gli angiomi racemosi, anche detti crisoidei o arterovenosi, presentano un groviglio di vene e arteriole, e hanno sede particolarmente profonda.
Angiomatosi
La condizione di angiomatosi, tipica dell'infanzia, può compromettere strutture vitali e favorire l'insorgenza di CID. Come già specificato, la angiomatosi è una forma di emangioma multiplo, disseminato.
Linfangiomi e simili
Anche per i linfangiomi sono descritte forme capillari e cavernose, similmente quindi alle forme dei vasi sanguiferi. I linfangiomi, peraltro, contemplano anche delle forme cistiche, o igromi cistici. L'igroma cistico è presente alla nascita, e probabilmente deriva da dilatazioni di caverne linfangiomatose; sedi preferenziali a capo, collo, mesentere, cute. Microscopicamente si osservano spazi vascolari rivestiti da cellule endoteliali, privi di eritrociti.
Le linfangiomatosi sono rare condizioni in cui si realizza una distribuzione diffusa di linfangiomi.
Le linfangiomiomatosi consistono nella proliferazione di fibrocellule muscolari lisce dentro i vasi linfatici e nei linfonodi. La sede è retroperitoneale, ad arrivare al parenchima polmonare. Sono principalmente colpite donne giovani, e ciò fa supporre un certo ruolo per i fattori endocrini.
Angiosarcomi
Gli angiosarcomi sono tumori vascolari maligni. Sono anche detti emangioendoteliomi maligni, o linfangioendoteliomi maligni.
Sedi preferenziali sono cute di capo e collo, parti molli, mammella, organi profondi (fegato, milza, ossa, cuore). Possiamo descrivere, tra gli angiosarcomi, una forma cutanea, una forma mammaria, una forma epatica; inoltre viene descritta una forma particolare, detta sarcoma di Stuart-Treves.
Forma cutanea. Macroscopicamente gli angiosarcomi sono rappresentati da focolai emorragici tendenti alla confluenza e alla ulcerazione cutanea. Al microscopio si osservano numerosi spazi vascolari irregolari, ripieni di sangue, più cellule endoteliali a diverso grado di atipia. Al microscopio elettronico si osservano i corpi di Weibel-Palade, che sono formazioni tubulari, presenti anche negli endoteliociti di aspetto normale. La immunoistochimica evidenza la presenza di vimentina e dell'Ag associato al fattore VIII. La neoplasia può metastatizzare per via ematogena o linfatica.
Sarcoma di Stuart-Treves. Insorge in seguito a linfedema cronico. Questa condizione può verificarsi all'arto superiore (ad es., dopo una mastectomia radicale) o all'arto inferiore (ad es., dopo una linfoadenectomia radicale inguinale per metastasi), dopo 5-10 anni dall'intervento. Macroscopicamente si presentano noduli multipli dermici e sottocutanei confluenti e ulceranti la cute.
Forma della mammella. Ad elevato grado di malignità, rapidamente progressivo, è mortale nel 90% dei casi entro 2 anni. Si riscontrano al microscopio focolai multipli angiomatosi anche in sede intralobulare (nelle forme benigne solo in sede interlobulare).
Forma epatica. Accertata la correlazione con l'assunzione di thorotrast e con l'esposizione al cloruro di polivinile.
Sarcoma di Kaposi
Anche detto "sarcoma idiopatico multiplo pigmentato cutaneo", è classicamente descritto come frequente in Africa, mentre in Europa si verifica sporadicamente. Colpisce l'età giovane media, e ha una sede cutanea, mucosa, viscerale, linfonodale. Macroscopicamente si osservano noduli dermici rosso-violacei o brunastri per lo più alle estremità inferiori. Al microscopio, invece, si osservano ammassi di capillari, con fibroblasti, periciti, cellule a capacità fagocitica (positivi all'emosiderina e al fattore VIII), macrofagi, plasmacellule, linfociti. Il decorso dell'affezione è lento, richiedendo mediamente 8-13 anni per estrinsercarsi, ed è fatale solo nel 10-20% dei casi. Raro, regredisce spontaneamente. Nei soggetti HIV+, la forma ha invece un decorso rapido, più aggressivo; la sopravvivenza è inferiore ai 24 mesi.
Tumore glomico
In realtà è quasi sempre benigno. Unico, si manifesta con dolori lancinanti in seguito ad esposizione ad acqua fredda o calda, oppure di notte. La sede è nel sottocutaneo o nel derma profondo, e molto frequentemente in sede sottoungueale.
Macroscopicamente il diametro è uguale o superiore a 1 cm, ben delimitato, e istologicamente risulta composto da cellule muscolari lisce che si sono trasformate in cellule glomiche; hanno nuclei rotondi e citoplasma chiaro, senza glicogeno. Si riscontrano inoltre ammassi di capillari, stroma mixoide o ialinizzato. La forma non delimitata prende il nome di glomangioma, e possiede diversi vasi dilatati.
Emangiopericitoma
Si tratta di una neoplasia che origina dalla proliferazione dei periciti di Zimmermann. Colpisce l'età adulta, e ha sede alle estremità inferiori, pelvi e retroperitoneo. Dalle dimensioni variabili, ha una buona delimitazione. Al microscopio si osservano formazioni vascolari rivestite internamente da endotelio normale, e circondate da periciti stipati fittamente. Esiste in forma benigna, potenzialmente maligna e francamente maligna.
La forma infantile (emangiopericitoma infantile) ha piccole dimensioni (fino a 5 cm), è tutto sommato benigno, ha una struttura multilobata. Le metastasi sono possibili solo se il tumore raggiunge elevate dimensioni, e in tal caso si localizzano a polmone e scheletro.
tumori sinoviali
I tumori sinoviali contemplano il sarcoma sinoviale e il tumore a cellule giganti delle guaine tendinee.
Tumore a cellule giganti delle guaine tendinee
Anche detto "tenosinovite villonodulare pigmentata", colpisce in età media e si localizza a dita, articolazioni del piede, caviglia, ginocchio. Esiste in forma nodulare o diffusa, extra-articolare.
Ha una crescita lenta, e un diametro che raggiunge tutt'al più i 3-4 cm. Al taglio mostra una superficie a colore giallo-bruno. Al microscopio si riscontra una modesta cellularità, e queste cellule possono essere rotondeggianti, poligonali oppure fusate; si riscontrano anche cellule giganti con molti nuclei centrali, e cellule schiumose a tipo xantico. Lo stroma può essere scarso o abbondante, ad aspetto fibroso.
Sarcoma sinoviale
Rappresenta, come frequenza, il 10% dei tumori maligni delle parti molli. Frequente in adolescenti, giovani adulti, sempre maschi. Ha sede nelle aree para-articolari in particolar modo agli arti, mentre è più raro a capo, collo, tronco.
Il sarcoma sinoviale può essere di grandi dimensioni, e in tal caso risulta circondato da pseudocapsula, oppure di piccole dimensioni, e in tal caso ha un aspetto solido o gelatinoso, con aree emorragiche o necrotiche, di colore grigio-roseo, è infiltrante e mal delimitato. Microscopicamente si distinguono due tipi cellulari; le cellule epiteliomorfe, rare, e le cellule fusate.
Il sarcoma può essere bifasico o monofasico. Monofasico significa che è composto in prevalenza da cellule epiteliomorfe oppure fusate. Bifasico invece significa che vi sono formazioni epiteliali e strutture formate da cellule voluminose con abbondante citoplasma. In questo citoplasma c'è un materiale colorabile come le mucine epiteliali e proteine dei filamenti intermedi di cheratina. Nel sarcoma bifasico sono frequenti i focolai di metaplasia pavimentosa con perle cornee, e la componente fusocellulare disposta a vortice, con scarso citoplasma e filamenti di vimentina (rara la cheratina).
Al microscopio elettronico le cellule epiteliomorfe presentano desmosomi, mentre le cellule fusate sono collegate da giunzioni tipo "zonula adherens".
Lo stroma può essere scarso o abbondante, e avere un aspetto mixoide, pseudo-osteoide o con focolai di calcificazione.
Metastasi vengono date nel 50-60% dei casi, e le sedi più frequenti sono polmoni, linfonodi regionali e midollo osseo. La sopravvivenza media a 5 anni è del 35-40%. La forma a prognosi peggiore è quella monofasica a cellule fusate.
TUMORI DEI NERVI PERIFERICI
Classificazione
La grande novità di questi tumori è che si classificano in benigni e maligni. Finalmente una cosa diversa! Le forme benigne contemplano lo schwannoma benigno, il neurofibroma, la neurofibromatosi, e atri. Le forme maligne, invece, contemplano lo schwannoma maligno, il tumore di Triton e il neuroepitelioma periferico.
Forme benigne
Schwannoma benigno. E' un tumore benigno, a lenta crescita, capsulato, che origina da un nervo (periferico o cranico) del sistema nervoso autonomo.
L'età di massima frequenza è tra 20 e 50 anni, e le sedi sono capo, collo, superfici flessorie delle estremità, mediastino posteriore, retroperitoneo. In realtà queste ultime due localizzazioni sono meno frequenti rispetto alle altre. Il diametro è superiore a 5 cm.
Da un punto di vista microscopico possiamo descrivere due quadri diversi, che però a volte coesistono. Il "tipo A" presenta cellule fusate, nuclei ovali disposti parallelamente, secondo gli assi maggiori; i nuclei possono essere disposti a palizzata, a vortici, o anche in altri modi, ma comunque con regolarità. Il citoplasma è eosinofilo, eventualmente a margini sfumati, e vi sono noduli stromali ialinizzati. Il "tipo B" presenta cellule fusate disposte invece in modo casuale, e una matrice lassa contenente aree infiammatorie e zone microcistiche.
Una varietà particolare di schwannoma è il tumore benigno a cellule granulari, un tempo considerato ad origine muscolare (e pertanto un mioblastoma).
Esso si presenta come un nodulo indolente, unico; età più frequente 30-60 anni, in donne. La sede può essere nel derma, nel sottocutaneo, a lingua, in sottomucose, alla muscolatura (liscia o striata), a laringe, bronchi, stomaco, vie biliari. Macroscopicamente è un focolaio mal delimitato di colorito giallo-brunastro, dal diametro massimo di 3 cm. Microscopicamente si osserva un ammasso di cellule rotondeggianti, il cui nucleo è piccolo e centrale, e il citoplasma è ampio, pieno di granuli eosinofili PAS+ e Masson+. Nuclei e citoplasma contengono la proteina S-100, i granuli citoplasmatici hanno proteine mieliniche, enolasi neurone-specifiche, proteine di filamenti di vimentina. Sono assenti proteine proprie della muscolatura striata e la proteina acida fibrillare gliale.
In sede linguale la neoplasia produce spesso una reazione pseudoepiteliomatosa dell'epitelio di rivestimento, diagnosticata erroneamente come carcinoma spinocellulare invasivo.
Neurofibroma. E' una lesione non capsulata, ma ben circoscritta. Se asportato completamente, non recidiva. Nella matrice intercellulare si ritrova collagene e componenti mucoidi e mixoidi. Macroscopicamente è possibile reperire il tronco nervoso che entra ed esce dalla neoplasia. Da un punto di vista microscopico, sono presenti cellule allungate, disposte in fasci separati da collagene e mucopolisaccaridi acidi; i nuclei sono ondulati o a virgola, ed intensamente colorabili.
Si riscontra la presenza di proteina S-100, mentre c'è assenza di neurofilamenti, proteina acida fibrillare gliale, enolasi neurone-specifica.
Neurofibromatosi. Detta anche malattia di von Recklinghausen, ha una frequenza di 1/3000, ha una ereditarietà autosomica dominante, con alta penetranza. Vi è una forma periferica e una centrale o acustica. Le caratteristiche importanti sono:
a) macchie cutanee color caffellatte;
b) amartomi pigmentati dell'iride;
c) neurofibromi multipli cutanei e profondi delle parti molli, che compaiono durante l'infanzia e l'adolescenza;
d) deformità scheletriche.
Nella forma centrale i neurofibromi sono presenti nel SNC, e c'è associazione con astrocitomi, meningiomi, ependimomi.
Da un punto di vista macro e microscopico i neurofibromi sono analoghi a quelli sporadici, oppure possono essere di tipo plessiforme; cioè il processo proliferativo può iniziare dal perinevrio, nel quale sono presenti cellule fusate. Da ricordare, infine, che queste neoplasie benigne possono avere trasformazione sarcomatosa, a tipo di schwannoma maligno.
Mixoma delle guaine tendinee. Forse è una variante di neurofibroma; la sede è sottocutanea o dermica, e l'età colpita è quella infantile. Vi sono caratteristici lobuli di sostanza mixoide inglobanti cellule isolate o raggruppate in nidi. Sono frequenti dopo traumi o interventi chirurgici; sono espressione di proliferazione disordinata di fascetti nervosi (neuroma traumatico da amputazione).
Forme maligne
Schwannoma maligno. Nel 50% dei casi si realizza in soggetti con neurofibromatosi, conclamata o mista. L'età prevalente è 30-50 anni, e si presenta coma una massa aggettante da un nervo, il cui diametro è inferiore a 5 cm, e con fenomeni emorragici e necrotici. Microscopicamente è simile al fibrosarcoma, ma in questo caso c'è un maggiore disordine strutturale, vi sono cellule con aspetto contorto, nuclei a virgola. Presenza focale di proteina S-100, presenza di vimentina. Stroma di aspetto mixoide.
Gli schwannomi maligni non insorti in corso di malattia di von Recklinghausen hanno prognosi migliore. Poco frequenti le metastasi per via linfatica, è invece frequente la disseminazione lungo lo stesso nervo, anche in senso centripeto, fino allo spazio subaracnoideo), e sono anche frequenti le recidive.
Tumore di Triton Può esistere in forma benigna o maligna. La forma maligna è in realtà uno schwannoma maligno con componente rabdomiosarcomatosa; c'è positività per desmina e mioglobina citoplasmatiche. Una sua variante è lo schwannoma maligno epitelioide, con una cellularità che simula un carcinoma o un melanoma maligno.
Neuroepitelioma periferico. Microscopicamente simile al neuroblastoma, compare in qualunque età e può insorgere in nervi periferici.
paraganglioma
Il paraganglioma origina dalla neurocresta. I paragangli appartengono ad un sistema che comprende la midollare surrenalica, i paragangli brachiomerici (corpo carotico, ganglio giugulo-timpanico), i paragangli intravasali, i paragangli aorto-simpatici. Le sedi dove un paraganglioma può insorgere sono queste appena citate, oppure in sede viscerale.
Le forme di paragangliomi più frequenti sono:
a) p. giugulo-timpanico;
b) p. vagale;
c) p. mediastinico;
d) p. retroperitoneale (ad origine dell'organo di Zuckerland);
e) chemodectoma (o p. non cromaffine).
Microscopicamente si osservano isolotti di cellule rotonde circondate da complessa vascolarizzazione, nuclei con cromatina grossolana, citoplasma amfofilo-granulomatoso o eosinofilo. Queste ultime, in realtà, sono anomalie cellulari (nucleari e citoplasmatiche) non indicative di malignità.
Una differenza va fatta tra le forme cromaffini e quelle non cromaffini. Le seconde dimostrano una fluorescenza indotta dai vapori di formalina su materiale non fissato. Questa fluorescenza è verde per le catecolamine, e gialla per la serotonina. Inoltre, vi sono granuli di secrezione neuroendocrina e capacità di ridurre i sali d'argento.
I paragangliomi sono maligni quando insorgono in sede retroperitoneale.
mesoteliomi
I mesoteliomi (tumori a origine mesoteliale) possono essere pleurici, pericardici, peritoneali.
Mesoteliomi pleurici
I mesoteliomi pleurici originano dal mesotelio, che può differenziarsi in senso epiteliale o connettivale. Distinguiamo mesoteliomi primitivi e mesoteliomi secondari. Ci sbarazziamo subito di questi ultimi; i mesoteliomi secondari derivano da propagazione linfatica di carcinomi polmonari, mammari, gastrici, ovaie/utero, reni. Macroscopicamente si individuano come reticolo biancastro a maglie lievemente rilevate (carcinosi endolinfatica), oppure come placche o noduli sporgenti, oppure ancora come estese infiltrazioni a piastrone. I mesoteliomi primitivi, invece, possono essere localizzati (forma nodosa) oppure diffusi.
Mesotelioma localizzato o nodoso. E' caratterizzato da un lento accrescimento, dal fatto di essere capsulato, a volte peduncolato. Le dimensioni sono variabili (si possono avere, ad es., delle forme giganti come il tumore della pleura viscerale di Klemperer); asportato, non dà recidive né metastasi. Frequentemente può comparire un focolaio emorragico. La consistenza del tumore è fibrosa. Alla superficie di taglio appare a colorito bianco-grigiastro, con aree emorragiche, e cavità cistiche contenenti liquido chiaro. Al microscopio si notano fasci regolari di cellule fusate, stroma collageno più o meno denso con aree scleroialine, e possibile presenza di aree mixoidi; in tal caso darebbe l'impressione di un fibrosarcoma, ma il suo andamento è in tal caso benigno. L'istogenesi di questo tumore è controversa: potrebbe originare dal tessuto areolare sottopleurico, oppure dai mesoteli.
Mesotelioma diffuso. Il tumore ha oggi una discreta frequenza; colpisce per lo più maschi tra 40 e 70 anni, e vi è evidente correlazione con esposizione all'asbesto.
Ha una fase preclinica prolungata, fino a dare dolori localizzati, difficoltà nel respiro, versamento pleurico con alti valori di acido jaluronico. Questo mesotelioma può seguire una pleurite cronica recidivante fibroplastica.
Macroscopicamente si realizza un notevole ispessimento della pleura (2-5 cm), aumento della durezza, aspetto bianco, lardaceo; il polmone - classicamente - è murato nella cotenna mesoteliale (il tumore è anche chiamato tumore a corazza), e il versamento - che si riforma facilmente dopo una toracentesi - è imponente. Il tumore può estendersi alla pleura parietale; i due foglietti possono aderire e fondersi, e il tutto può simulare un fibrotorace.
Microscopicamente si possono osservare due diverse componenti:
a) aspetto epiteliale simil-carcinomatoso, con isole, cordoni, alveoli di cellule rotondeggianti con citoplasma acidofilo e nucleo vescicoloso nucleolato, insieme a formazioni tubulari rivestite da cellule cubiche o cilindriche;
b) aspetto connettivale, con stroma ricco di cellule fusate con fisionomia fibromatosa o sarcomatosa.
L'istogenesi di questo tumore è dal mesotelio (il duplice aspetto istologico è tipico). La diagnostica si avvale della misurazione dei livelli acido jaluronico (elevati) e di indagini ultrastrutturali.
Per queste ultime, si osservano lunghi microvilli con positività al ferro colloidale, neolumi (?) nelle cellule con microvilli, presenza di membrana basale, granuli di glicogeno intracitoplasmatico, cellule fibroblastoidi (a forma fusata con microvilli), positività del CEA.
Il comportamento biologico è - come tutti sanno - maligno, e la sopravvivenza si attesta intorno a 4- 12 mesi. Poveracci!
Mesoteliomi del pericardio
Anche questi mesoteliomi possono essere primitivi o secondari.
Mesoteliomi del pericardio primitivi. Possono mostrare aspetti differenti; ad esempio possono avere un aspetto carnoso, di nodosità multiple, a consistenza duroelastica, oppure avere un aspetto di masse mollicce, con aree di colliquazione necrotica. Gli aspetti sono diversi anche sul piano microscopico. Infatti è possibile riscontrare aspetti a tipo fibrosarcomatoso, e aspetti a tipo epiteliomorfo, con elementi epiteliali ammassati in strutture simil-ghiandolari. La diversità degli aspetti dipende dalle molteplici capacità evolutive dell'epitelio celomatico di rivestimento pericardico.
Mesoteliomi del pericardio secondari. Derivano da diffusione ematogena o linfatica da carcinoma polmonare o gastrico (più spesso polmonare). In caso di carcinoma gastrico, ma che di carcinoma esofageo, si osservano strie di carcinosi endolinfatica con essudazione siero-emorragica nel cavo pericardico (si parla di pericardite neoplastica). In caso di melanomi, invece, si possono osservare masse mollicce associate a pericardite emorragica.
Mesoteliomi del peritoneo
Anche per quanto attiene i mesoteliomi del peritoneo, è possibile individuarne di primitivi e di secondari.
Forme primitive. Originano dal mesotelio o dal connettivo sottomesoteliale. I tumori mesoteliali possono essere papillomi o mesoteliomi. Analizziamo i mesoteliomi.
Macroscopicamente si osservano placche o noduli simili a quelli tubercolotici, biancastri, isolati o estesi, con peritonite sierofibrinosa a carattere emorragico. Microscopicamente possono essere individuate quattro forme:
a) tubulo-papillare. Ci sono papille tappezzate da cellule cubiche o cilindriche, cellule con acido jaluronico, stroma abbondante;
b) indifferenziata. Ci sono cellule rotonde o poliedriche disposte in cordoni solidi compatti, stroma poco abbondante;
c) mista. Con associazione di proliferazione epiteliale indifferenziata con cellule fibro-sarcomatose;
d) sarcomatosa: Con cellule fusate.
La diffusione può avvenire per contiguità, per via linfatica, per via ematica (ovaio, fegato).
I tumori che originano dal connettivo sottomesoteliale comprendono forme benigne e forme maligne. I tumori benigni hanno sede in omento o mesentere, e sono rotondeggianti o peduncolati, consistenti o mollicci; microscopicamente possono avere l'aspetto di fibromi, fibromiomi, lipomi, leiomiomi, linfangiomi (per lo più cistici). I tumori maligni, invece, possono essere linfangiosarcomi, emangiosarcomi, fibrosarcomi, mixosarcomi.
Tra i tumori primitivi del peritoneo è anche contemplato il tumore carcinoide del peritoneo.
Forme secondarie. Hanno una discreta frequenza. Macroscopicamente può esserci:
a) carcinosi miliare, con noduli piccoli e numerosi;
b) carcinosi nodosa, con noduli grossi; frequente in omento e mesentere;
c) carcinosi infiltrante massiva, di tipo scirroso o gelatinoso.
Coesiste essudato endoperitoneale sieroso.
Descriveremo brevemente come, tra le forme secondarie, lo pseudomixoma benigno del peritoneo, e la splenosi del peritoneo.
Lo pseudomixoma benigno del peritoneo si presenta come masse gelatinose libere o incapsulate in connettivo vascolare e con cellule giganti. Può derivare dalla rottura nel cavo peritoneale di un cistoma pseudomucinoso dell'ovaio oppure da raccolte mucose asettiche dell'appendice. Può essere localizzato o generalizzato. La sua patogenesi prevede due teorie differenti: potrebbe trattarsi di una peritonite da corpo estraneo (teoria infiammatoria) oppure di metastasi da impianti mucosecernenti (teoria neoplastica).
La splenosi del peritoneo consiste nella presenza, sulla superficie peritoneale, di noduli sessili, numerosi, piccoli, aventi la struttura della milza con la polpa rossa (molto frequentemente) e la polpa bianca, insieme a capsula e ilo vascolare. Bellissimo! Suggestivo! E' vero che esiste, non è stata inventata. La sua genesi potrebbe prevedere una milza sovrannumeraria, o disturbi di sviluppo, o un seguito di rottura traumatica della milza.
MAMMELLA
mastiti
Mastiti acute
Sono anche dette puerperali, e colpiscono molto frequentemente donne primipare durante le prime 4 settimane.
Causa dell'infiammazione è l'infezione da Stafilococco aureo, la cui penetrazione è compiuta attraverso soluzioni di continuo del capezzolo, o dell'areola, e la cui propagazione può essere canalicolare o linfatica. Canalicolare, cioè attraverso i dotti galattofori prima, i dotti interlobulari poi, e infine nel parenchima ghiandolare; si realizza una flogosi acuta diffusa, cui segue una fase necrotico-colliquativa che coinvolge le strutture acino-duttali, con formazione finale di un ascesso mammario o di un flemmone. La propagazione linfatica, invece, si realizza attraverso i vasi periduttali, che portano al tessuto interstiziale della ghiandola.
Gli ascessi mammari sono suddivisi in subareolari (nella regione sottostante il capezzolo), ghiandolari (interessano uno o più lobi e possono aprirsi in un grosso dotto galattoforo o all'esterno) e retromammari (al davanti della fascia pettorale). La progressione di una mastite acuta può consentire una suppurazione, con formazione (ovviamente) di tessuto di granulazione e tessuto cicatriziale, oppure una guarigione senza reliquati.
Mastiti croniche aspecifiche
Possono far seguito alle mastiti acute non suppurative. Si osserva una infiltrazione linfocitaria del connettivo peri e intralobulare con ispessimento dello stroma. Nelle infezioni suppurative, si può raggiungere un elevato grado di durezza per quanto riguarda la reazione connettivale, e ciò può trarre in inganno e far diagnosticare un carcinoma! Soprattutto se consideriamo la reazione linfoghiandolare satellite. Ma chiaramente, con una puntura esplorativa, in caso di processo di origine suppurativa, verrà fuori il pus.
Un tipo particolare è la mastite plasmacellulare. Si tratta di una affezione infiammatoria non infettiva, monolaterale. L'esordio può essere acuto o subacuto. L'esordio acuto, in realtà, è ben manifesto, con tumefazione consistente a limiti indistinti, aderente alla cute del capezzolo e con linfoadenopatia (anche in questo caso si impone una diagnosi differenziale con i carcinomi). Questa forma di mastite si realizza frequentemente in donne che hanno allattato diversi figli, in menopausa.
Al microscopio si osserva una notevole infiltrazione plasmacellulare, ma anche di linfociti, istiociti e cellule schiumose, con proliferazione dell'epitelio di rivestimento dei dotti galattofori. Si formano cellule giganti tipo Langhans, senza però necrosi caseosa. Anche i tessuti periduttali sono frequentemente infiltrati da elementi flogistici.
Ectasia dei dotti mammari
Si tratta di una lesione flogistica che colpisce i dotti escretori extralobulari. In donne, con più di 45 anni e gravidanze multiple. L'etiopatogenesi offre a considerare due interpretazioni. Secondo una di esse, la stasi del secreto provocherebbe dilatazione dei dotti e flogosi. E quindi è importante la stasi del colostro, un microtraumatismo da suzione, un deficit nello sviluppo dei dotti galattofori. Seconda un'altra interpretazione, la dilatazione sarebbe secondaria ad una flogosi periduttale.
Una ectasia in fase acuta consente il riscontro microscopico della dilatazione di alcuni dotti lattiferi nella zona profonda del capezzolo. Questi si trasformano in cisti dal diametro di 2-4 mm; nel lume c'è materiale amorfo lipidico e macrofagi schiumosi. Tali cisti sono rivestite da epitelio cubico o appiattito, e sono anche presenti ocrociti o fluorociti; queste ultime due parolacce indicano cellule a citoplasma con pigmento ceroide. Queste cellule sono posizionate tra l'epitelio, oppure nel lume, oppure nel connettivo pericistico, e sono di natura macrofagica o mioepiteliale. Sempre relativamente alla presenza di queste cisti, bisogna dire che c'è intensa flogosi pericistica e periduttale, e interessamento retrogrado dei dotti extralobulari più piccoli.
Quando le lesioni flogistiche sono più severe si possono avere due aspetti caratteristici. Uno di essi è a tipo comedomastite. Si realizza in questo caso una notevole dilatazione dei dotti, un ispessimento delle pareti e, nel lume dei dotti, presenza di sostanza amorfa acidofila, cristalli di acidi grassi e numerose cellule schiumose lipidofore. Intorno ai dotti si realizza una flogosi granulomatosa con cellule flogistiche. Clinicamente si osserva fuoriuscita di materiale sieroematico dal capezzolo, e retrazione del capezzolo stesso.
Un'altra possibilità di presentazione per la forme più severe è a tipo mastite granulomatosa. In tal caso la flogosi si estende nel connettivo interlobulare, con ascessi; si formano granulomi composti da cellule epitelioidi, cellule giganti tipo Langhans, cellule mononucleate, polimorfonucelati. Anche, qui, assenza di caseosi. I processi riparativi portano a riduzione (o anche a scomparsa) della flogosi, che viene sostituita da un tessuto fibroso periduttale ad anello, continuo o discontinuo. E ciò fa parlare di mastite obliterante quando il quadro è particolarmente marcato. Clinicamente si osserva retrazione del capezzolo.
Mastiti croniche specifiche
Tubercolosi. Rara la localizzazione mammaria. Comunque, si realizza in donne giovani e gravide, e la via di infezione è ematica, o linfatica retrograda dai linfonodi ascellari, o per contiguità. La tubercolosi mammaria contempla quattro forme anatomiche: la forma miliare, la nodosa (o lobulite tubercolare), l'ascesso freddo (può aprirsi all'esterno fistolizzando; sulla cute la lesione avrà i margini sottominati), la galattoforite tubercolare. Ad ogni modo, si realizza un granuloma con necrosi caseosa.
Sifilide. Si realizzano tre differenti aspetti. Il sifiloma primario del capezzolo o dell'areola è un nodulo che va incontro ad ulcerazione a margini rilevati duri, con adenopatia ascellare. Il sifiloma secondario si manifesta con flogosi interstiziale della ghiandola mammaria; regredisce spontaneamente. Il sifiloma terziario, infine è una nodosità dapprima consistente, poi rammollita, e che infine ulcera; è la cosiddetta gomma luetica, ma è rara. Non è associata a linfoadenopatia.
Noccioline finali. Altri quadri di mastiti croniche specifiche sono la actinomicosi (dà luogo a seni fistolosi multipli) e la sarcoidosi (in realtà molto rara).
Liponecrosi mammaria
Si tratta di una necrosi del tessuto adiposo in sede mammaria. Essa può essere causata da pratiche mediche (punture esplorative, biopsie, exeresi), da traumi pregressi passati inosservati, oppure essere l'espressione di una panniculite disseminata idiopatica.
La liponecrosi mammaria è un'affezione a decorso subdolo; si manifesta con un nodulo di 2-3 cm di diametro, duro, indolente, aderente a cute (la cute assume un aspetto "a buccia d'arancia"). Frequentemente è localizzato nella regione areolare.
Il reperto macroscopico anatomo-patologico evidenzia, nelle fasi recenti, una zona opaco-biancastra e zone emorragiche, se la forma segue un trauma. In seguito si realizza una possibile evoluzione colliquativa centrale, con formazione di una pseudocisti a contenuto oleoso, e circondata da connettivo fibroso denso. Nei casi vecchi si osservano depositi di calcio nelle pareti cistiche.
Microscopicamente si osserva una necrosi focale degli adipociti, con rottura della membrana citoplasmatica; il materiale lipidico liberato viene fagocitato dai macrofagi, e ciò causa una reazione flogistica, con comparsa di tessuto di granulazione; si tratta del famoso granuloma lipofagico, che è simile a quello della TBC. In pratica si osserveranno vacuoli otticamente vuoti attorno ai quali si dispongono grossi macrofagi a citoplasma schiumoso, plasmacellule e cellule giganti tipo "da corpo estraneo". Se la lesione è datata, invece, si osserveranno neoformazioni connettivali, calcificazione del materiale necrotico e possibile sostituzione, parziale o totale, da parte di tessuto fibroso dell'area di liponecrosi.
mastopatia fibroso-cistica
Si tratta di una condizione che viene - o è stata - anche denominata iperplasia fibroso-cistica, o displasia mammaria, o è stata chiamata col nome di alcuni autori, come Reclue o Schimmelbosh. Ma tutto ciò poco importa.
E' la contemporanea presenza di alterazioni di tipo regressivo (cisti, fibrosi) e di tipo proliferativo (iperplasia epiteliale, adenosi), con partecipazione di elementi epiteliali e connettivali.
La mastopatia fibroso-cistica è spesso bilaterale, e ha sede nei dotti terminali e nelle strutture epiteliali dei lobuli; l'età più colpita è la giovane-media (massimo 40 anni), e si apprezza come una serie di nodosità palpabili. Questa malattia deriverebbe da uno squilibrio di crescita epiteliale e connettivale, collegato a fattori ovarici (persistente stimolazione estrogenica).
Può esistere in forma diffusa o circoscritta; la lesione ha una elevata consistenza, limiti mal definiti, presenza di cisti macroscopiche e microscopiche; queste possiedono un liquido di vario aspetto, e, se sottoposte a pressione, mostrano l'aspetto di cupola azzurrognola. La parete interna di queste formazioni è liscia e lucente.
Questa lesione è molto "divertente" per i patologi, che possono così esprimere al massimo (o quasi) la loro cervellotica e disumana Weltanshauung (non saprei trovare parola migliore). Difatti adesso analizzeremo le caratteristiche delle varie "cose" che questa malattia presenta al microscopio.
Cisti
Le cisti sono tra gli elementi distintivi della mastopatia fibroso-cistica. Esse sono inquadrabili come cisti semplici o cisti con metaplasia apocrina.
Cisti semplici. Le cisti più grandi sono rivestite da un singolo strato di cellule, cubiche o piatte per compressione; le cisti più piccole sono rivestite da un epitelio cubico spesso, disposto in due strati.
3-4 microcisti occupano di solito un lobulo intero. Esse possono rimanere tali oppure fondersi tra loro e formare della macrocisti. Le microcisti devono essere differenziate con gli esiti dell'involuzione fisiologica del lobulo (laddove comunque sono di più e presentano un connettivo lasso intralobulare); le macrocisti devono essere differenziate dalla ectasia dei dotti extralobulari (dove in realtà compaiono fibre elastiche attorno alle cisti). Le cisti macroscopiche (o macrocisti) sono avvolte da una reazione flogistica cronica, con plasmacellule e macrofagi (talvolta schiumosi). L'epitelio si mostra discontinuo, desquamato e talvolta c'è trasformazione fibrosa del connettivo pericistico.
Cisti con metaplasia apocrina. In questo tipo di cisti le cellule appaiono provviste di caratteristiche istochimiche e ultrastrutturali simili a quelle delle cellule apocrine. Il rivestimento di queste cellule è un epitelio cilindrico, con cellule a citoplasma eosinofilo, pallido, e nucleo parabasale, ipercromatico. Sono presenti pluristratificazioni con propaggini nel lume della cisti. Nel citoplasma vi è costante presenza di granuli glicolipidici (detti di Lendrun). Questo tipo di cisti originerebbe come la risultante di un fenomeno inizialmente ostruttivo, oppure come una neoformazione di dotti su base ormonale.
Adenosi e varianti
Si tratta dell'iperplasia dei duttuli terminali. Vi sono, principalmente, due tipi di adenosi: l'adenosi a dotti ciechi e l'adenosi sclerosante. L'etiologia prevede due ipotesi: la prima è che si tratti di variazioni iperplastiche di strutture lobulari pre-esistenti; la seconda è che si tratti di formazioni "de novo" a partenza dai dotti lattiferi.
Adenosi a dotti ciechi. Si distinguono forme organoidi e forme non organoidi. Tra le forme organoidi, con componente connettivale, vi sono la adenosi semplice, la adenosi microcistica, la adenosi nodulare.
Nella adenosi semplice si realizzano formazioni acinose a cellule epiteliali ipertrofiche con citoplasma colonnare che presenta propaggini apicali di secrezione; è la metaplasia colonnare di Boaser. Nelle fasi conclamate vi è un elevato numero di tali cellule, con dilatazione degli acini, numerose cellule mioepiteliali e ancora una gran quota di connettivo lasso intralobulare. Nella adenosi microcistica c'è una maggiore dilatazione delle strutture epiteliali intralobulari rivestite dallo stesso tipo di cellule che formano propaggini pseudopapillari; il lobulo risulta formato da 4-5 cisti rivestite da cellule colonnari. La adenosi semplice e microcistica presentano anche delle varianti non organoidi, che hanno caratteristiche un po' diverse e vanno sotto il nome di adenosi microghiandolari; queste forme possono trovarsi associate a carcinoma mammario.
La adenosi nodulare presenta focolai nodulari, costituiti da connettivo speciale che contiene formazioni acinari; tali formazioni acinari sono rivestite da cellule epiteliali a citoplasma ridotto con propaggini apicali secretorie, e cellule mioepiteliali nello strato più esterno. Queste ultime cellule perdono rapporti con la membrana basale e le cellule epiteliali e si dispongono in fascetti nel tessuto interstiziale ("trasformazione mioide"). Nelle fasi conclamate i noduli hanno poche strutture acinari, a lume ristretto, e molte cellule rotondeggianti e fusate; inoltre, vi sono mitosi a carico di cellule epiteliali e mioepiteliali.
C'è una variante di adenosi caratterizzata da elevato numero si strutture acinari e un abbondante connettivo lasso, edematoso; si chiama iperplasia fibro-adenomatosa.
Adenosi sclerosante. Qui si ha una proliferazione stromale e fibrosi, con distorsione delle strutture tubulari. Interessa l'età giovane-adulta (25-35 anni). Clinicamente è un nodulo simil-tumorale, palpabile, dal diametro di 2,5 cm. Non è capsulato, ha un colorito bianco-grigiastro, consistenza sostenuta. Alla superficie di taglio dimostra dei limiti poco definiti e una configurazione a vortice.
Microscopicamente si apprezzano delle formazioni nodulari che, contrariamente a quanto avviene per il carcinoma, sono più cellulari al centro e hanno un connettivo fibroso periferico. Vi sono cellule mioepiteliali con trasformazione mioide, e vi può essere elastosi. Caratteristiche costanti, comunque, sono l'aspetto nodulare, organoide, l'assenza di pleomorfismo, la doppia differenziazione epiteliale e mioepiteliale.
Iperplasia epiteliale
E' la proliferazione di cellule epiteliali nei dotti e lobuli presistenti. Vi sono iperplasie duttali e iperplasie lobulari. Da notare che i due tipi di iperplasia raramente coesistono in uno stesso lobulo.
Iperplasia duttale. Si realizza all'interno dei dotti terminali e segmentari extralobulari. Origina dalla zona di passaggio tra parte intra e parte extralobulare dei dotti terminali. E' anche detta epiteliosi, per il fatto di essere quasi sempre diffusa. Vi è sempre un certo grado di distensione delle strutture interessate per dilatazione del lume. Nei dotti si riscontrano aspetti di accrescimento solido con piccoli spazi vuoti periferici, aspetti fenestrati per la presenza di spazi vuoti, aspetti tipo propaggini pseudo-papillari, linguiformi che aggettano nel lume.
Le cellule sono monomorfe, simil-epiteliali, a contorni indefiniti, quasi sinciziali. I nuclei non hanno nucleoli e sono parzialmente embricati. Mancano aree di necrosi, emorragia, calcificazioni. Alla periferia sempre presenti cellule mioepitaliali.
Una variante dell'iperplasia duttale è detta epiteliosi infiltrante; qui si realizza una fibrosi delle aree centrali delle lesioni, in cui sono immerse piccole formazioni tubulari distinte. Talvolta, elastosi centrale. E' detta anche proliferazione papillare sclerosante, o adenosi con pseudoinfiltrazione.
Iperplasia lobulare. La proliferazione cellulare si realizza all'interno dei duttuli terminali (acini). Ha insorgenza autoctona, da cellule di riserva parabasali duttulari. Nello stato iniziale si realizza una proliferazioni di nidi di cellule tra cellule epiteliali e mioepiteliali; queste cellule sono piccole, e hanno un nucleo ipercromatico centrale; con la progressione si ha il coinvolgimento di tutti i duttuli. Allo stadio finale, infatti, si ha che il lobulo è aumentato di volume, molti duttuli sono dilatati e zaffati da cellule strettamente coesive tra loro, e sono evidenti bene le cellule epiteliali e le cellule mioepiteliali.
Esiste anche una iperplasia lobulare atipica, in cui le cellule hanno una scarsa coesione, limiti mal definiti, polimorfismo nucleare.
tumori della mammella
Tumori benigni
In genere insorgono in relazione ad una abnorme sensibilità locale agli stimoli ormonali; si classificano in fibroadenomi, papillomi, amartomi; poi c'è un'entità particolare, il cosiddetto tumore filloide.
Fibroadenomi. Molto frequenti, sono mobili, hanno consistenza sodo-elastica, hanno una estensione circoscritta e di solito sono unici; quando si è di fronte a più fibroadenomi con margini indistinti si parla propriamente di fibroadenomatosi.
I fibroadenomi colpiscono più frequentemente donne giovani, e si localizzano al quadrante superesterno della mammella; hanno lenta crescita in età fertile, per poi "regredire" con abbondante produzione di tessuto fibroso dopo la menopausa (in ciò si denota il ruolo degli ormoni nel mantenimento della lesione).
Macroscopicamente si presenta come un nodulo rotondeggiante, dal diametro di 1-3 cm; quando il diametro supera i 4 cm si parla di "fibroadenoma gigante". E' dotato di pseudocapsula e, al taglio, dimostra una superficie bianco-rosea, lucente. Al microscopio si osserva una proliferazione di connettivo mixoide e di tubuli epiteliali; talvolta vi sono reperti di iperplasia epiteliale (le cellule epiteliali, cioè, si dispongono su più strati) con metaplasia apocrina cellulare.
Esistono delle varietà anatomiche di fibroadenoma. Esse sono l'adenoma, il fibroadenoma intracanalicolare, il fibroadenoma pericanalicolare, e le forme miste.
L'adenoma è una proliferazione con scarsità/assenza di componenti connettivali; il fibroadenoma intracanalicolare è composto da canalicoli compressi e deformati in fessure per via dell'espansione del connettivo mixoide verso il lume; infine, il fibroadenoma pericanalicolare presenta un connettivo che è disposto concentricamente ai tubuli. Si tratta, per quest'ultima forma, di una variante con spiccata agressività e che risulta da aggredire chirurgicamente.
Tumore filloide. Si tratta di una neoformazione mammaria a carattere fondamentalmente benigno, in cui il tessuto neoformato si esplica attraverso ramificazioni complesse, evidenti in sezione. C'è iperplasia dei fibroblasti, ma anche delle cellule connettivali; quest'ultimo particolare è stato scoperto con studi di microscopia elettronica. Le proliferazioni connettivali sono compresse, e al taglio sgusciano fuori "a cavolfiore". Il tumore ha una elevata velocità di crescita, e può dare recidive locali dopo asportazione incompleta; è stata descritta qualche trasformazione sarcomatosa, e per questo motivo è stato anche chiamato "cistosarcoma filloide". In realtà sia l'aspetto cistico che l'aspetto sarcomatoso sono rarissimi.
Papillomi. Si sviluppano a livello dei dotti mammari, dove l'epitelio è su due strati; lo strato più superficiale contiene elementi cellulari cubici, mentre lo strato basale contiene cellule mioepiteliali. Vi sono diversi tipi di papilloma: il papilloma solitario, il papilloma multiplo, l'adenoma del capezzolo.
Il papilloma solitario si sviluppa sui dotti galattofori e sui dotti interlobulari; il diametro può arrivare al cm. Può dimostrare un sanguinamento per rottura delle papille da cui è formato. In corrispondenza del papilloma, che è una formazione ostruente, può formarsi, a monte, una cisti; questa cisti avrà contenuto ematico e sarà circondata da connettivo sclerotico. Biologicamente questo papilloma è assolutamente benigno.
I papillomi multipli si sviluppano in più dotti e, preferibilmente, alla periferia del parenchima ghiandolare, per cui non sono apprezzabili palpatoriamente; biologicamente sono "pericolosi", hanno cioè una relativa scarsa benignità.
L'adenoma del capezzolo, formazione benigna, è un piccolo nodo sottoepidermico, costituito dalla proliferazione dell'epitelio dei dotti in prossimità dello sbocco alla cute; anzi, in quella sede possono osservarsi piccole ulcerazioni dell'epidermide.
Amartomi. Benigni, si presentano clinicamente come fibroadenomi, con i quali vengono spesso confusi; in realtà si tratta di neoformazioni caratterizzate da sovrabbondanza di elementi fisiologicamente presenti in detta sede, e nulla di più.
Tumori maligni
I tumori maligni della mammella (primitivi) possono essere di natura carcinomatosa o sarcomatosa. I sarcomi, in realtà, sono molto rari. Dei carcinomi, invece, ce n'è quanti se ne vuole; alcune forme sono più frequenti, altre meno. Per questo motivo qui si parlerà solo di carcinomi.
Poiché nella popolazione femminile il carcicoma mammario è molto diffuso, da parecchio tempo si sono aperti degli studi atti ad individuare le precancerosi, cioè delle lesioni, dimostrabili istologicamente, le quali sono "più che associate" allo sviluppo di tumore. Esiste cioè un elenco di lesioni la cui dimostrazione in una paziente aumenta il rischio - di quella paziente rispetto ad una paziente nella quale la lesione non c'è - di sviluppare un carcinoma mammario. Ora, questa "possibilità" è espressa in statistica con una misura, OR, "Odds Ratio". Un OR = 4, ad es., indica un rischio quadruplicato ( x 4 ). E allora, per quelle precancerosi che hanno un OR fino a 4 i più Autori indicano un semplice monitoraggio periodico, mentre per lesioni che hanno un OR da 4 a 12, e, a maggior ragione, superiore a 12, un intervento chirurgico preventivo. In realtà, proprio queste lesioni più a rischio sono di meno frequente osservazione.
Aspetti morfologici. La gran maggioranza dei carcinomi avrebbe origine dai duttuli pre-terminali, situati alla giunzione tra i dotti interlobulari ed i canalicoli che formano il lobulo mammario. La proliferazione delle cellule neoplastiche causerebbe una successiva dilatazione dei piccoli dotti, che assumono notevoli diametri e assomigliano a dotti interlobulari. Per questo motivo si giustifica la dizione "carcinoma duttale" per alcuni carcinomi mammari.
Le sedi più frequenti sono, nell'ordine, il quadrante superolaterale, il centrale, il superomediale, l'inferomediale, l'inferolaterale. Il carcinoma mammario clinicamente rilevabile ha il diametro di 1 cm, e può avere due aspetti, quello di un nodo duro retraente e quello di un nodo encefaloide, oltre a forme miste. Questi due aspetti diversi sono dovuti al differente sviluppo di connettivo fibroso. E' indicato l'uso del criostato durante l'intervento, anche perché delle forme benigne possono simulare macroscopicamente forme maligne.
In 3/4 dei casi la morfologia è quella del nodo duro retraente (detto scirrro); esso risulta avere una compattezza soda, lignea. E' fisso e resistente alla sezione, che risulta netta, grigio-rosea e asciutta; a volte può mostrare macchie bianche e giallastre per via di fatti necrotici, con successiva calcificazione ad aspetto comedonico. Il contorno non è netto. Il nodo è detto retraente per via della presenza di miofibroblasti, che conferiscono alla cute sovrastante un aspetto "a buccia d'arancia".
Il nodo encefaloide in sezione ha l'aspetto "di cervello e midollo"; ha un colorito grigio-roseo, con screziature emorragiche, consistenza molle e contorni regolari.
Quando si realizza una infiltrazione diffusa dei vasi linfatici, con esteso turgore della mammella, si parla di carcinoma infiammatorio.
Microscopicamente nella gran parte dei casi si tratta di un adenocarcinoma, quindi di una neoplasia ad aspetto istologico ghiandolare. Una classificazione "tradizionale", ma grossolana e imprecisa suddivide i carcinomi mammari in CR in situ (o non invasivi), CR invasivi e forme combinate rare. Sia i CR in situ che i CR invasivi possono essere duttali o lobulari; in realtà questi due aggettivi indicano non una diversa istogenesi, ma solo la conformazione e lo sviluppo neoplastico. Cioè non è che i CR duttali derivino da cellule dei dotti!
CR in situ duttale (o CR intraduttale). Si tratta di forme palpabili e rilevabili alla mammografia. I realizza una proliferazione di cellule epiteliali nella parete dei dotti, con rispetto della membrana basale; la parete dei dotti è ispessita e dilatata e il lume può essere compresso (aspetto solido). Talvolta il cordone centrale va in necrosi (aspetto cordonico), oppure i cordoni possono formare lumi secondari (aspetto cribriforme) o, ancora, possono formare digitazioni (aspetto papillare); ovviamente sono anche possibili aspetti associati. Le cellule sono globose, policlonali; vi sono atipie nucleari. A seconda del grado di differenziazione sono diverse le possibilità di recidiva.
CR in situ lobulare. Si tratta di una forma il cui riscontro è occasionale (accanto a lesioni macroscopiche). E' sviluppato negli estremi a fondo cieco dei duttuli, e le cellule sono globose, chiare, con una certa omogeneità; raramente si osservano mitosi. La parete di tali formazioni si dilata, il lume si riduce e si riempie. Anche in questo caso c'è il rispetto della membrana basale. Vi si osservano cellule mioepiteliali residue addensate alla membrana basale. Le dimensioni di questa neoformazione possono essere molto piccole, e il fattore di rischio è pari a 11.
CR invasivo duttale. Rappresenta l'80% dei casi di CR mammario; si osservano irregolari cordoni e nidi di cellule epiteliali, disomogenee, con nuclei grandi e mitosi. Talora si osservano calcificazioni ghiandoliformi. Spesso c'è abbondante connettivo (aspetto scleroso) fibroso o ialino. In periferia, prolungamenti cordoniformi di cellule epiteliali in tessuto adiposo; i margini delle neoplasia possono apparire quindi stellati, ma anche netti e rotondeggianti.
CR invasivo lobulare. A prognosi grave, costituiscono il 10% dei casi. Le cellule proliferano nel connettivo, sono piccole, uniformi e in fila indiana. Hanno vacuoli citoplasmatici ripieni di muco (si tratta in realtà di introflessioni della membrana cellulare), che fanno parlare di "ghiandola unicellulare".
Forme meno frequenti di CR mammario sono il CR midollare, il CR mucinoso, ed altre forme, che poi verranno semplicemente elencate.
CR midollare. Nodo circoscritto, a margini arrotondati, è formato da masse e cordoni di cellule epiteliali, uniformi e voluminose; lo stroma possiede linfociti. Il tumore ha scarsa invasività.
CR mucinoso. Area molle, a limiti indistinti. In sezione vi è materiale gelatinoso, colloso, translucido. Microscopicamente si osserva muco con cellule più o meno aggregate cilindro-cubiche o poligonali a diversa dislocazione: a nidi (anziani) o ghiandolare. Scarsa è la tendenza alla metastatizzazione.
Forme rare sono il CR tubulare (dalla rara metastatizzazione), i CR papilloforo invasivo, adenoideo-cistico, argirofilo, secretorio giovanile, apocrino.
Considerazioni finali. Fattori prognostici che hanno un peso nella valutazione di una neoplasia mammaria sono le metastasi linfonodali, la sede, il volume, gli emboli carcinomatosi vascolari, il tipo istologico, il grado di malignità, il numero di mitosi, lo sviluppo in contemporanea con la gravidanza. Si tratta di cose pressoché ovvie e comunque conosciute o intuibili.
I tumori mammari possono essere multicentrici: 2 lesioni compresenti ma diversamente sviluppate. La diffusione locale è lungo i dotti, per via linfatica e per via ematica.
Metastasi vengono date ai linfonodi ascellari e mediastinici anteriori; i primi interessati sono a livello del piccolo pettorale, poi al gran pettorale e quindi in sede ascellare. Altre metastasi sono date poi al midollo osseo e a epidermide, polmoni, cervello. La stadiazione prevede l'utilizzo del classico TNM.
La patologia neoplastica maligna mammaria contempla anche i sarcomi e i linfomi. Ma basti questo.
Malattia di Paget del capezzolo
Si tratta di una affezione flogistica, eczematosa, ragadiforme e a volte ulcerosa dell'area capezzolo-areola. Il vetrino presenta una epidermide con grandi cellule a citoplasma diafano e nucleo atipico, conserate neoplastiche (cellule di Paget); però queste cellule non infiltrano lo stroma. Probabilmente sono cellule migrate lungo i dotti, e poi all'interno dell'epidermide (i dotti lattiferi mostrano comunque un CR intraduttale). L'atteggiamento che bisogna avere in questi casi è, comunque, quello verso un tumore maligno. E' possibile che si realizzi un "Paget extramammario" (ghiandole sudoripare!) con lo stesso significato.
TUMORI ENDOCRINI DEL PANCREAS
Sotto questa infelicissima dizione vanno considerati i tumori, cioè i processi neoplastici, che coinvolgono la funzione endocrina del pancreas. Considereremo l'adenoma insulare e il carcinoma insulare.
adenoma insulare
È il più frequente tumore benigno del pancreas, e ha una età di insorgenza dai 35 ai 60 anni. Può essere solitario o multiplo, e la localizzazione più frequente è alla coda (70%); un minor numero di casi interessa la testa. Altre volte si manifesta in sede aberrante. Il suo diametro può essere compreso tra 1 e 10 centimetri.
Macroscopicamente si presenta rotondeggiante, capsulato, ben delimitato, e omogeneo in superficie di taglio. Microscopicamente presenta cellule polimorfe, grandi, con nuclei grossi e chiari, ma soprattutto delle isole cellulari, anastomizzate tra loro in intimo contatto con capillari (cosiddetta rosetta, cioè disposizione attorno ad un capillare).
Carcinoma insulare
Costituisce il 10% dei tumori insulari. Macroscopicamente si osservano degli aspetti che, in verità, non sono patognomonici di malignità in questo caso. Essi sono la molteplicità dei nodi, la loro incompleta demarcazione, e la recidiva.
Microscopicamente si osservano diverse cosette interessanti; innanzitutto, c'è l'infiltrazione neoplastica della capsula connettivale peritoneale, vi sono mitosi numerose e atipiche, c'è un grado di polimorfismo cellulare. Le cellule presentano grossi nucleoli. Si riscontra invasione dei vasi linfatici e sanguigni, con presenza di estesi focolai di necrosi.
Poiché le componenti insulari - in senso di cellule - sono parecchie, si distinguono diverse varietà di carcinoma, in rapporto al tipo istofunzionale.
Tumori a cellule beta. Microscopicamente possono osservarsi strutture solide (varietà midollare) oppure nidi o cordoni separati da sinusoidi (varietà trabecolare). Le cellule sono scarsamente granulate, per via dell'elevato contenuto in proinsulina. Si riscontra amiloidosi dello stroma e depositi di sali calcici. Il tumore dà luogo alla caratteristica triade di Whipple, con crisi ipoglicemiche a digiuno, tasso glicemico a digiuno inferiore a mg 50 per mille, pronta scomparsa della crisi in seguito a somministrazione di glucosio.
Tumori a cellule alfa. Si caratterizza per la negatività delle cellule neoplastiche al test di Gomori. Microscopicamente si realizza una raccolta in strutture solide. Il tumore si manifesta con iperglicemia, glicosuria, diabete mellito, dermatite eritematosa-papulosa con glossite (da deposizione tissutale di Triptofano).
Tumori a cellule G. Si tratta della sindrome di Zollinger-Ellison tipo II. Si realizza la triade: tumore insulare, ipersecrezione acida gastrica, ulcera peptica grave in sede atipica (zona distale del digiuno). La neoplasia dà metastasi ai linfonodi regionali. Microscopicamente si osservano isole o cordoni di cellule rotondeggianti. La conferma che si tratti di un tumore endocrino viene ottenuta tramite l'utilizzo di opportune colorazioni (grimelius, ematossilina al Pb), e l'esclusione del fatto che non sia un tumore a cellule viene dalla negatività del Gomori. La genesi avviene per deficit della differenziazione delle cellule pluripotenti.
Tumori a cellule D. Si osservano granuli tondeggianti privi di alone [sic, NdR]. Il tumore dà luogo a ipoinsulinemia, ipoglucagonemia, diarrea, acloridiria. Negatività con grimelius [aridàje, NdR] e sieri antigastrina.
Tumori a cellule D1 o VIPomi. Anche detti "sindrome di Verner-Morrison". Si manifestano con diarrea acquosa (si parla di colera pancreatico), ipocaliemia, ipo/acloridria. La sindrome è conosciuta anche con l'acronimo anglosassone WDHA, cioè "Watery Diarrhea, Hypokaliemia Acloridria".
Altri. Altri istotipi possibili sono le cellule PP e le cellule che producono serotonina; questi ultimi danno luogo ai tumori con il ridicolo nome di serotoninomi.
Ovviamente tutte le volte che si parla di "citologia" si intende "tecnica citologica"; povero idioma!
E' bellissimo pensare che l'apparecchio, con le sue braccia, spinga un liquido verso un filtro. e mentre rotea!
|
