Le traslocazioni che coinvolgono i cromosomi del sesso devono essere considerate separatamente perché il cromosoma X è capace di inattivarsi
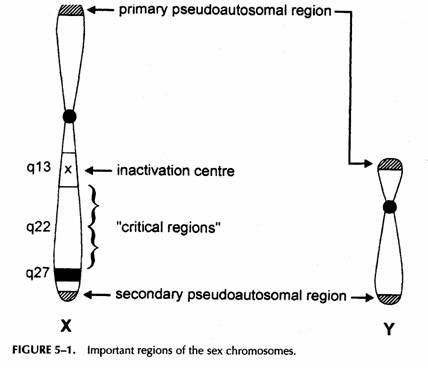
PATTERN DI INATTIVAZIONE NELLA FEMMINA PORTATRICE DI UNA TRASLOCAZIONE BILANCIATA X-AUTOSOMICA.
Vi sono due cromosomi con riarrangiamenti contenenti materiale dell'X. Un segmento riarrangiato della X contiene XIC e può essere inattivato, l'altro non lo contiene e sarà quindi sempre attivo.
In un portatore di traslocazione X-autosomica solitamente viene inattivata la X intatta.
Questo meccanismo funziona in ¾ delle eterozigoti. In un quarto dei casi il meccanismo fallisce e sopravvivono alcune cellule che sono funzionalmente disomiche per tratti dell'X. La disomia causa effetti fenotipici.
Solo cellule con piccole disomie sopravvivono e quindi questo fenomeno è più frequente con punti di rottura Xp od Xq distali.
Il fenotipo può anche essere dovuto ad inattivazione (spread della inattivazione) del segmento autosomico traslocato
Lo stato di in attivazione può essere studiato con il replication banding che distingue l'X attiva dalla inattiva.
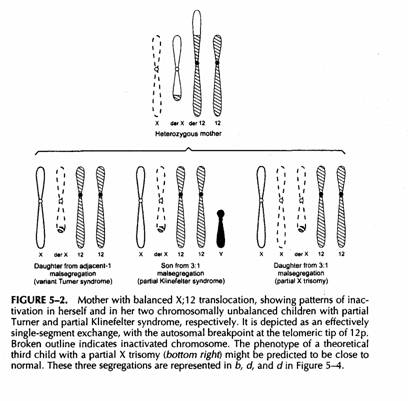
PATTERN DI INATTIVAZIONE IN FIGLI SBILANCIATI DI UNA DONNA PORTATRICE BILANCIATA (i maschi portatori sono sempre sterili).
In un figlio sbilanciato di un portatore di traslocazione X-autosomica l'effetto dello sbilanciamento può essere mitigato dalla inativazione selettiva della X anormale. In questo caso anche la trisomia parziale per il segmento traslocato può essere corretta sempre che lo spread della inattivazione sia completo.
Se tuttavia Il segmento traslocato che origina dalla X non contiene XIC non potrà essere inattivato e vi sarà una disomia funzionale parziale per la X ed una monosomia autosomica

Traslocazioni a singolo segmento
a) il segmento singolo traslocato è quello della X.
Una figlia che riceva la X con la delezione (in questo caso una Xq-) in luogo della X normale, o come cromosoma addizionale avrebbe una forma parziale di aneuploidia dei cromosomi sessuali
Una figlia con 46,X,-X, +der(X) da segregazione adiacente-1 avrebbe una variante Turner
Un figlio con trisomia terziaria 47,XY,+der(X) avrebbe un Klinefelter incompleto ed una figlia 47,XX,+der(X) una sindrome 47,XXX incompleta.
Nel caso illustrato concepimenti con 46,-12, + der(12) da segregazione adiacente-1 sarebbero disomici per una grande quantità di X e quindi non vitali.
Se il segmento X traslocato è però piccolo la disomia parziale dell'X potrebbe essere vitale
b) il segmento singolo traslocato è quello autosomico
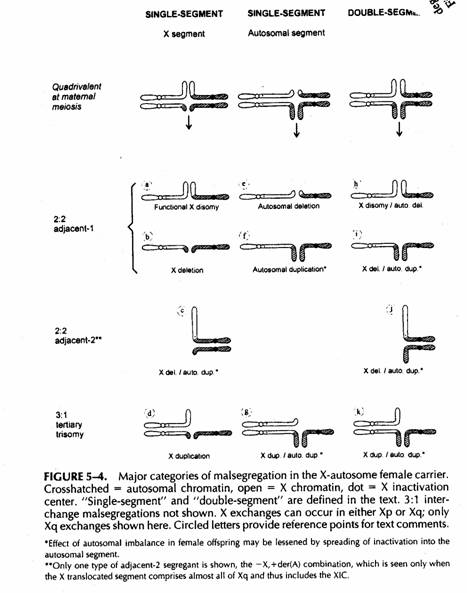
I concepiti sbilanciati per 2:2 sarebbero parzialmente trisomici o monosomici per il tratto autosomico traslocato (46,-A,+der(A) e 46,-X,+der(X))
Lo stato trisomico parziale nella femmina ha un fenotipo che può essere attenuato da spreading della inattivazione al segmento traslocato. Nel maschio questo non accade e la trisomia parziale non viene attenuata.
Lo stato di monosomia rimane identico come nel caso di una traslocazione non X-autosomica.
Scambio a doppio segmento
con una segregazione adiacente-1 ci possono essere effetti di una combinata disomia funzionale della X e monosomia autosomica, od una monosomia della X ed una trisomia autosomica.
Questi effetti nella femmina parzialmente trisomica 46,X,-X,+der(X) possono essere modificati dallo spread della inattivazione
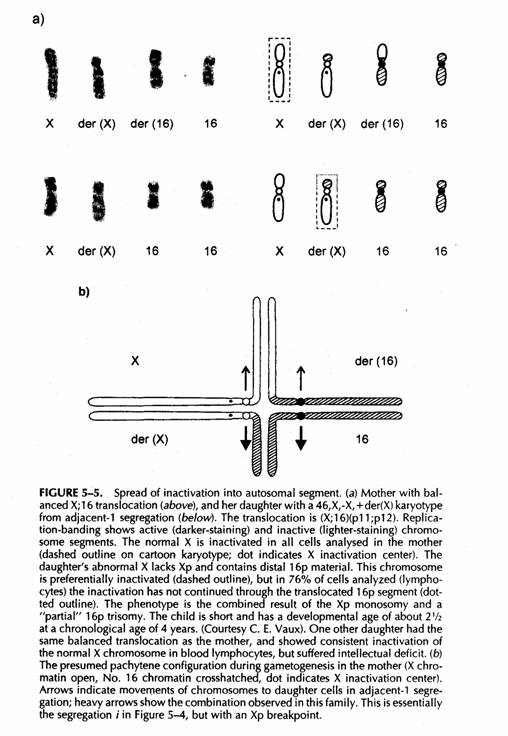
La stessa situazione col la Y (46,Y,-X.+der(X) con nullisomia Xp/trisomia 16p sarebbe letale in utero
Le altre segregazioni adiacenti-1 46,XX,-16, +der(16) non sarebbero correggibili e determinerebbero monosomia 16p/disomia funzionale Xp (incompatibile con la vita)
La segregazione adiacente-2 produce trisomia per molta parte di un cromosoma e monosomia per l'altro
Questi gradi enormi di sbilanciamento possono essere però accomodati in un concepito di sesso femminile.
P.es l' autosoma intatto e quello riarrangiato sono trasmessi assieme: 46,X,-X,+der(A). Se il tratto di X include XIC lo spread della inattivazione potrebbe correggere la trisomia autosomica. La concomitante monosomia X è vitale (Turner)
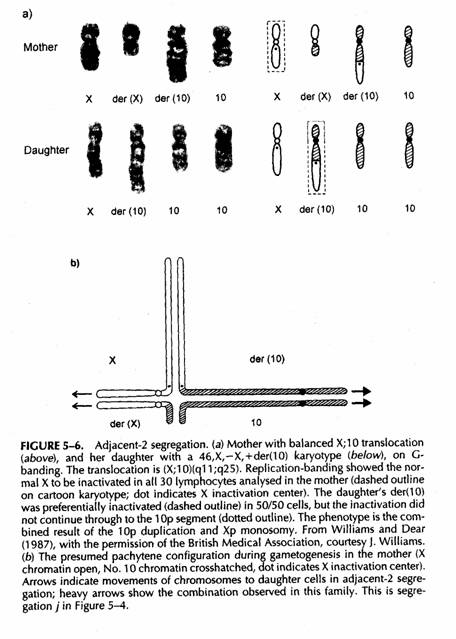
Vitalità sarebbe anche possibile se 46XX,-A, +der(X), dove der(X) contiuene XIC, il segmento autosomico contiene gran parte del genoma dell'autosoma e lo spread dell'inattivazione non coinvolge il frammento traslocato autosomico (l'autosoma deve avere un braccio corto geneticamente piccolo p.es 22)
Gli stessi criteri si applicano alla monosomia terziaria da segregazione 3:1
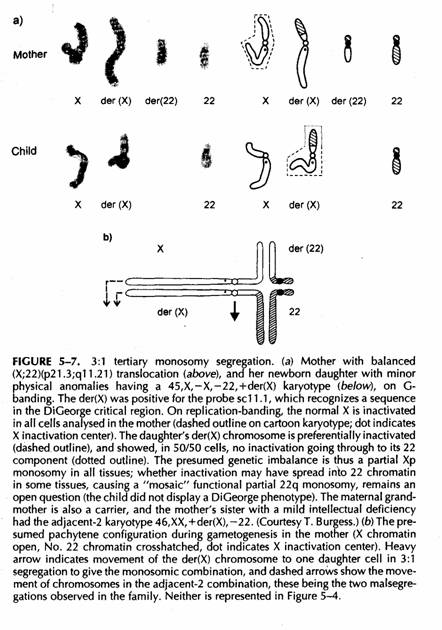
Il der(X) contiene gran parte dell'X ed anche del 22
Nella metà dei casi l'acrocentrico è il 15 e nell'altra il 22. spesso è familiare (maschi e femmine coinvolti ugualmente) senza conseguenze fenotipiche
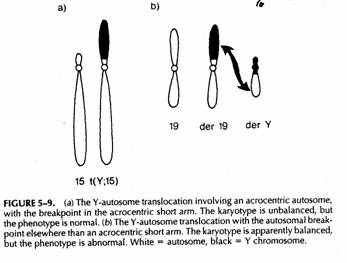
Raro e de novo apparentemente bilanciato. Azoospermia ipogonadismo, ritardo mentale
TDY viene a trovarsi su un altro cromosoma. Il soggetto ha 45 cromosomi e può apparentemente sembrare un 45,X
La traslocazione classica
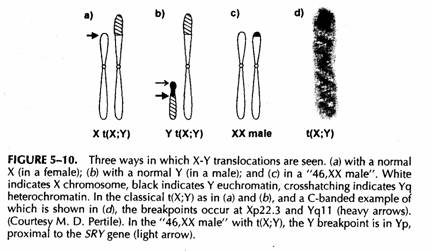
Tre forme differenti.
La forma classica ha rotture in Xp distale e Yq prossimale (delezione della regione distale dell'Xp.
Parziale monosomia X nella femmina, bassa, IQ normale e fertile
Parziale nullisomia Xp nel maschio, ritardo mentale e fenotipo da geni contigui Xp.
La maggioranza dei casi è familiare
80% dei maschi XX e 45,X
46,XYq- e scambio criptico Xq-Yq
de novo. disomia Xq distale con fenotipo grave.
traslocazione X-autosomica
Tutti i maschi e metà delle femmine sono infertili
Se la femmina è fertile ha un rischio sostanziale di avere figli sbilanciati dal 20 al 40%, con fenotipo da lieve (parziale Klinefelter o parziale monosomia X) a severo(disomia parziale X, aneuploidia autosomica non modificata dalla inattivazione)
LA TRASLOCAZIONE BILANCIATA VISTA IN PRENATALE
Il fenotipo può essere differente da quello di una madre anch'essa portatrice della traslocazione. In un quarto dei casi il fenomeno di inattivazione della X normale fallisce e l'individuo è disomico per tratti della X (fenotipo patologico). Quando la rottura è prossimale ad Xp22 e Xq28 il rischio è minore (lo sbilanciamento funzionale incompatibile con la vita)
Y-AUTOSOMICHE
Normale variante
Azoospermia ipogonadismo, ritardo mentale
Aumentato rischio di 45,X o 47,XXY
TRASLOCAZIONE CLASSICA X-Y
La forma classica ha rotture in Xp distale e Yq prossimale (delezione della regione distale dell'Xp).
Parziale monosomia X nella femmina, bassa, IQ normale e fertile
Ha il 50% di rischio di avere un figlio od una figlia con la stessa anomalia
Parziale nullisomia Xp nel maschio, ritardo mentale e fenotipo da geni contigui Xp.
La maggioranza dei casi è familiare e di derivazione meiosi maschile
80% dei maschi XX e 45,X
46,XYq- e scambio criptico Xq-Yq
de novo. disomia Xq distale con fenotipo grave.
|
