Cromatografia de gaze reprezinta acea tehnica cromatografica ce utilizeaza o faza mobila gazoasa si o faza stationara lichida sau solida. Este cea mai utilizata tehnica cromatografica, deoarece prezinta o serie de avantaje care îi confera o adaptabilitate deosebita la problemele care pot apare în timpul analizei amestecurilor complexe de componente. Astfel, prin utilizarea unui gaz drept faza mobila se realizeaza un transfer de masa rapid între faza mobila si faza stationara, ceea ce permite stabilirea echilibrelor de distributie de multe ori într-un interval scurt de timp si obtinerea unor separari eficiente corelate cu timp de analiza redus. Vâscozitatea scazuta a gezelor permite utilizarea unor coloane lungi si înguste si în consecinta cresterea numarului de talere teoretice. Faza mobila fiind un gaz inert, dupa separare componentele pot fi usor detectate deoarece faza mobila nu interfera la detectie. Daca este cazul, componentele separate pot fi analizate suplimentar prin metode spectroscopice cum ar fi spectrometria de masa sau spectrofotometria în IR.
Singurele limitari ale cromatografiei de gaze sunt determinate de volatilitatea prea scazuta sau instabilitatea termica a componentelor analizate. si aceste dezavantaje pot fi însa limitate, de exemplu prin transformarea compusilor respectivi în derivati care sa aiba volatilitate mai mare. Tehnica se numeste derivatizare si este larg utilizata în cromatografia de gaze.
În cromatografia de gaze, parametrii de retentie trebuie corectati pentru a tine cont de compresibilitatea fazei mobile. Din cauza acestei compresibilitǎti, volumul si debitul fazei mobile cresc în timpul trecerii prin coloanǎ. Aceste cresteri sunt cu atât mai mari cu cât raportul dintre presiunea la intrarea în coloanǎ pi si presiunea la iesirea din coloanǎ po este mai mare.
Pentru corectarea volumelor de retentie ca urmare a compresiei fazei mobile se defineste un factor de corectie numit factor de compresie, care se noteazǎ cu j si are valoarea datǎ de ecuatia:
![]()

![]() (48)
(48)
Ţinând cont de factorul de compresie, vom putea defini un volum de retentie corectat VRo si un volum de retentie net VN, conform relatiilor:
![]() (49)
(49)
![]() (50)
(50)
Un alt parametru de retentie caracteristic pentru cromatografia gaz-lichid este volumul de retentie specific Vg. El se defineste ca volumul de retentie net raportat la 0ºC si 1 g fazǎ sta 111i83b tionarǎ lichidǎ.
![]() (ml/g·ºK) (51)
(ml/g·ºK) (51)
unde WS este masa de fazǎ stationarǎ lichidǎ din coloanǎ (în grame), T este temperatura absolutǎ a coloanei, F debitul de fazǎ mobilǎ la iesirea din coloanǎ (în ml/sec), iar tR' este timpul de retentie ajustat al componentei (în secunde). Volumul de retentie specific este un parametru independent de aparat si de conditiile de lucru, caracteristic pentru solut si faza stationarǎ respectivǎ. Este asadar un parametru calitativ, care s-ar putea utiliza pentru identificarea unei componente, însǎ din pǎcate determinarea sa experimentalǎ este dificilǎ pentru cǎ depinde de factorul de compresie j.
Volumul de retentie specific se poate exprima si în functie de coeficientul de activitate si presiunea de saturatie a componentei, considerând cǎ în sistemul cromatografic ea se gǎseste într-o solutie extrem de diluatǎ. Relatia, dedusǎ pe baza legii lui Henry, este:
![]() (52)
(52)
unde R este constanta generalǎ a gazelor, MS masa molecularǎ a fazei stationare, po presiunea de saturatie a componentei la temperatura coloanei, iar γ coeficientul de activitate al componentei la dilutie infinitǎ în faza stationarǎ.
Coeficientul de activitate reprezintǎ o mǎsurǎ a interactiunii dintre moleculele componentei dizolvate în faza stationarǎ lichidǎ si moleculele fazei stationare.
Dacǎ considerǎm volumele de retentie specifice a douǎ componente care au picuri vecine si facem raportul lor, obtinem pe baza relatiei (51):

iar pe baza relatiei (52):
![]() Rezultǎ
deci:
Rezultǎ
deci:
![]() (53)
(53)
Relatia (53) ne aratǎ cǎ raportul dintre volumele de retentie specifice a douǎ componente cu picuri vecine este o mǎsurǎ a selectivitǎtii sistemului cromatografic pentru componentele respective. Aceastǎ selectivitate, exprimatǎ prin coeficientul de separare α, este determinatǎ de diferenta dintre presiunile de saturatie sau coeficientii de activitate ai componentelor. Asadar separarea a douǎ componente se va putea realiza în douǎ modalitǎti:
indicii de retentie
Cromatografia este o excelenta metoda de separare, dar care nu ofera nici un fel de informatii structurale despre componentele amestecului de analizat. Singurele informatii calitative obtinute în urma analizei cromatografice sunt reprezentate de timpii de retentie ai componentelor (sau volumele de retentie corespunzatoare), ceea ce este foarte putin în conditiile în care acest parametru depinde foarte mult de conditiile de analiza: temperatura, debitul fazei mobile, natura si concentratia fazei stationare, lungimea si diametrul coloanei. În plus, este foarte posibil ca mai multi compusi sa aiba acelasi timp de retentie în conditii de analiza identice. Asadar probabilitatea unor identificari calitative eronate este foarte mare daca ele se fac numai pe baza timpului de retentie.
În vederea realizarii unor analize cromatografice calitative mai exacte se folosesc urmatoarele metode:
- determinarea unor parametri de retentie relativi, care depind în masura mai mica de conditiile de analiza;
- utilizarea unor detectoare specifice, care ofera anumite informatii si despre structura substantelor analizate;
- cuplarea cu tehnici spectrometrice (spectrometrie de masǎ, spectrofotometrie în infrarosu), care ofera informatii structurale nemijlocite despre componentii separati din proba de analizat.
Dintre parametrii de retentie relativi, în analiza calitativa se utilizeaza retentia relativa si indicii de retentie Kovats.
Retentia relativa se defineste prin raportul dintre volumele de retentie specifice, sau timpii de retentie ajustati, al compusului analizat si al unui compus considerat standard pentru analiza respectiva, conform relatiei:
 (54)
(54)
Retentia relativa este dependenta de temperatura si de natura fazei stationare, dar independenta de celelalte conditii de analiza: lungimea coloanei, debitul de gaz purtǎtor, factorul de compresie si uneori de raportul fazelor, întrucât ele afecteaza în mod egal timpul de retentie ajustat al componentei de analizat si al standardului. Este indicat ca timpul de retentie al standardului ales sǎ nu difere foarte mult de cel al compusului analizat. De aceea nu exista un compus standard universal, el alegându-se pentru fiecare amestec de analizat în functie de conditiile concrete ale analizei respective. Însǎ în cazul unui anumit laborator, daca numarul compusilor care se analizeaza nu este foarte mare, este posibil sa se foloseasca tot timpul acelasi standard si sa se creeze o banca de date proprie de retentii relative care sa permita identificarea calitativa a compusilor respectivi.
S-a constatat cǎ pentru o serie omoloagǎ de compusi existǎ o relatie liniarǎ între numǎrul atomilor de carbon si logaritmul volumului de retentie. Aceastǎ relatie a fost folositǎ pentru a defini indicii de retentie Kovats. Kovats a propus folosirea n-alcanilor ca si compusi standard universali pentru analiza calitativǎ pe baza retentiei relative. Ei prezinta avantajul ca sunt usor accesibili si foarte stabili din punct de vedere chimic. Cu ajutorul lor au fost definiti niste parametri numiti indici de retentie si o scala a acestor indici de retentie pe care fiecare alcan are o valoare egala cu 100 x n, n fiind numarul de atomi de carbon al alcanului.
Indicele de retentie al unui anumit compus x se determina prin interpolare între indicii de retentie a doi alcani, cu n si reapectiv n+z atomi de carbon, alesi astfel încât timpul de retentie al compusului sa fie situat între cei ai alcanilor respectivi. Se utilizeaza relatia:
 (55)
(55)
Rezultǎ:
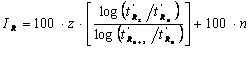
sau dacǎ tinem cont de faptul cǎ raportul timpilor de retentie ajustati reprezintǎ retentiile relative:
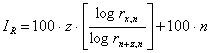 (56)
(56)
unde rx,n este retentia relativǎ a componentei fatǎ de alcanul n, iar rn+z,n este retentia relativǎ a celor doi alcani.
Indicii de retentie se determinǎ pe o anumitǎ faza stationara si la o anumitǎ temperaturǎ. Precizia determinarii creste atunci când eficienta de separare a coloanei este suficient de mare pentru a încadra compusul de analizat între doi alcani consecutivi ai seriei lor omoloage, cu alte cuvinte când z=1. Valorile indicilor de retentie pentru o serie de compusi pe diverse faze stationare au fost determinate si se gǎsesc si în literatura de specialitate. Cu ajutorul lor se poate aprecia calitativ posibilitatea separǎrii a 2 compusi pe o anumitǎ fazǎ stationarǎ. Se considerǎ cǎ o diferentǎ de cel putin 30 de unitǎti între valorile indicilor de retentie duce la o separare satisfǎcǎtoare, desi acest lucru nu este întotdeauna valabil.
Daca faza stationara este lichida, ea trebuie sa fie fixata pe un suport solid adecvat. Suportul are rolul de a retine faza stationara sub forma unui film subtire si uniform, permitând formarea unei interfete cât mai extinse între faza mobila si faza stationara. În acelasi timp însa, în cromatografia de repartitie suportul nu are voie sa interactioneze cu componentele probei, nici fizic nici chimic.
Conditiile pe care trebuie sa le îndeplineasca un suport utilizat în cromatografia gaz-lichid pe coloane cu umplutura sunt urmatoarele:
suprafata specifica situata în intervalul 1-20 m2/g;
inertie chimica;
rezistenta mecanica si stabilitate termica ridicata;
dimensiuni uniforme ale porilor.
Deoarece nu exista un suport ideal care sa corespunda tuturor cerintelor, selectarea suportului se face în primul rând pe baza suprafetei specifice si inertiei chimice.
Contrar aparentelor, un suport nu este cu atât mai bun cu cât suprafata sa specifica este mai mare, deoarece la suprafete specifice foarte mari nu toata suprafata ar fi acoperita de filmul de faza stationara lichida si ar exista pericolul adsorbtiei componentelor. Din acest motiv se aleg în general suporturi cu suprafata specifica situata între 1-7 m2/g. Acestor suprafete le corespund pori cu diametre situate între 0,5 -5 μm. Un alt parametru important este volumul intern al porilor, care pentru un suport bun trebuie sa fie în jur de 1 ml/g.
O alta caracteristica importanta este granulatia suportului. Diametrul mediu al particulelor si uniformitatea acestora au o influenta importanta asupra eficientei coloanei cromatografice. Un suport bun trebuie sa fie constituit din particule cât mai uniforme, de preferinta sferice si cu diametre de valori apropiate. În acest mod creste eficienta de separare a coloanei, prin scaderea înaltimii echivalente a talerului teoretic, în conformitate cu ecuatia Van Deemter (scade valoarea coeficientilor λ si γ care depind de iregularitatea marimii particulelor si a spatiilor dintre particule). Tot din ecuatia Van Deemter rezulta ca reducerea diametrului mediu al particulelor de suport determina cresterea eficientei coloanei. Aceasta reducere este însa limitata, deoarece dimensiuni prea mici ale particulelor ar duce la pierderi de presiune mult prea mari în coloana, deci presiunea gazului purtator la intrarea în coloana ar trebui sa fie mult mai mare, ceea ce ar genera o multime de probleme.
Granulatia suportului se exprima în mod traditional în mesh, care reprezinta numarul de ochiuri al sitelor folosite pentru obtinerea fractiunii respective, raportat la 1 inch (2,54 cm). De exemplu, un suport de 80-100 mesh este constituit din particule care au trecut prin sita cu 80 de ochiuri/inch dar au fost retinute de sita cu 100 de ochiuri/inch. Acestei granulatii îi corespund diametre ale particulelor între 0,13-0,15 mm.
Proprietatile mecanice ale suportului sunt de asemenea importante deoarece sfarâmarea particulelor, mai ales în timpul depunerii fazei stationare sau umplerii coloanei, determina scaderea eficientei colanei.
3.3.1.1. Dezactivarea suporturilor
Cea mai importanta caracteristica a unui suport al fazei stationare este inertia chimica. Deoarece nu exista materiale naturale corespunzatoare conditiilor impuse unui suport cromatografic care sa fie si suficient de inerte, este necesara modificarea suporturilor în sensul cresterii acestei inertii chimice.
Majoritatea materialelor utilizate drept suporturi sunt pamânturi de diatomee, care din punct de vedere chimic sunt derivati de acid polisilicic. Unele dintre aceste materiale, mai ales cele care contin oxizi de fier si aluminiu, pot avea activitate catalitica si exista pericolul sa catalizeze descompunerea fazei stationare, mai ales daca se lucreaza la temperaturi ridicate. Aceasta activitate catalitica este în general corelata si cu activitate de adsorbtie superficiala, care are drept urmare adsorbtia unei parti din componenta de analizat si aparitia picurilor cu cozi. Reducerea capacitatii catalitice si activitatii superficiale a suportului se face prin îndepartarea asa-numitelor centre active de pe suprafata acestuia. Exista trei tipuri de asemena centri activi:
- centre bazice, care determina formarea picurilor cu cozi în cazul solutilor cu caracter acid, de exemplu acizi carboxilici sau fenoli. Existenta acestor centre se datoreste oxizilor metalici (de Fe, Al) care se gasesc în structura suportului. Eliminarea lor se face prin spalarea acida a suportului, având loc concomitent si reducerea activitatii catalitice.
- centre acide, care formeaza picuri cu cozi în cazul compusilor bazici ca aminele sau anumiti compusi heterociclici. Eliminarea acestora se realizeaza prin spalare bazica.
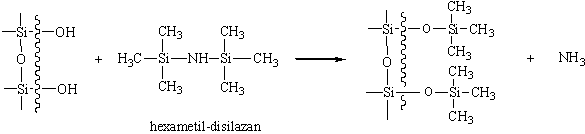
- centre de legaturi de hidrogen, care determina formarea picurilor cu cozi în cazul compusilor neutri, dar care pot da legaturi de hidrogen: alcooli, aldehide, esteri, eteri, etc. Existenta acestor centre active se datoreste existentei unor grupari silanolice (Si-OH) pe suprafata suportului. Neutralizarea acestora se realizeaza prin modificarea chimica a gruparilor respective, în special prin transformare în eteri sililici (Si-O-Si). Aceasta operatie se numeste silanizare, iar reactivii de silanizare cei mai utilizati sunt clor-trimetil-silanul, diclor-dimetil-silanul, hexametil-disilazanul si altele. Este important ca acesti compusi sa aiba toxicitate cât mai redusa si volatilitate ridicata, pentru ca reactivul de silanizare în exces sa poata fi usor eliminat la terminarea reactiei. Pentru exemplificare, reactia cu hexametil-disilazanul poate fi reprezentata schematic astfel:
Modul în care a fost tratat suportul se indica în denumirea comerciala a suportului, alaturi de natura si granulatia suportului. De exemplu, AW înseamna spalat acid (acid washed), iar DMCS înseamna ca suportul a fost silanizat cu dimetil-diclor-silan, asa cum se poate vedea în exemplul din Figura 3.1:
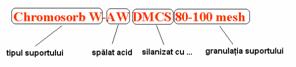
Figura 3.1. Semnificatia denumirii a suporturilor
Suporturile cele mai mult utilizate sunt cele pe baza de Kieselghur sau pamânt de diatomee, care contin acid polisilicic în forma amorfa hidratata, având structura poroasa si continând cantitati variabile de oxizi metalici de Fe, Al, Mg, Ca, Na, K. Aceste suporturi sunt cunoscute sub diferite denumiri comerciale, cele mai utilizate fiind cele de tip Chromosorb. Acestea se gasesc în mai multe varietati, cele mai importante fiind:
Chromosorb P (pink) are culoare roz-rosiatica si suprafata specifica situata în intervalul 4-6 m2/g. Are avantajul ca poate fi încarcat cu o mare cantitate de faza stationara lichida (pâna la 25-30%) si are rezistenta mecanica ridicata. Nu este însa suficient de inert chimic. Din acest motiv se utilizeaza mai ales la separarea substantelor nepolare.
Chromosorb W (white) are culoare alba si suprafata specifica mai mica, de 1-2 m2/g. Din acest motiv se poate încarca cu o cantitate mai mica de faza stationara lichida de cel mult 15%. Are inertie chimica buna, însa este friabil. Are aplicabilitate generala, fiind utilizat la separarea tuturor claselor de compusi, atât polari cât si nepolari.
Chromosorb G are suprafata specifica si mai mica, de numai 0,5 m2/g si se poate încarca cu maxim 5% faza stationara. Are însa avantajul de a combina inertia chimica a tipului W cu rezistenta mecanica a tipului P.
Exista suporturi si de alta natura, dar care sunt mult mai putin utilizate. Asemenea suporturi sunt cele pe baza de polimeri sintetici (de exemplu teflon), sticla si altele.
Conditiile pe care o faza stationara lichida trebuie sa le îndeplineasca sunt:
Sa fie un bun solvent pentru componentele probei analizate, însa solubilitatea acestor componente sa fie diferentiata;
Sa fie practic nevolatila la temperatura de lucru a coloanei (presiunea sa de vapori sa fie mai mica de 0,1 mm Hg);
Sa fie inerta din punct de vedere chimic;
Sa aiba o stabilitate termica cât mai ridicata.
Desi alegerea fazei stationare este foarte importanta pentru realizarea unei analize bune, nu exista o regula generala pe baza careia sa se poata face aceasta alegere, doar reguli semiempirice. Ea este usurata daca se cunosc cât mai multe date despre compusii analizati, în special structura chimica si temperatura de fierbere. O asemenea regula empirica este cea a similaritatii de structura, ceea ce înseamna ca faza stationara trebuie sa aiba o structura asemanatoare cu componenta analizata. Astfel, hidrocarburile se vor separa cel mai bine pe faze stationare parafinice (nepolare), iar substantele polare pe faze stationare polare.
Relatia (53) ne-a aratat ca selectivitatea separarii a doua componente depinde de presiunile de saturatie ale acestora si de coeficientii lor de activitate. Coeficientii de activitate reflecta interactiunile dintre faza stationara si componentele analizate. Aceste interactiuni pot fi de urmatoarele tipuri:
Interactiuni nepolare, care se datoresc unor forte de dispersie (de tip London) si se manifesta între moleculele unor substante nepolare. Deoarece aceste interactiuni sunt slabe, separarile unor asemenea compusi se vor produce în conformitate cu presiunile lor de vapori, adica ordinea de elutie din coloana va fi data de punctele de fierbere ale componentelor analizate.
Interactiuni dipol-dipol, care se stabilesc între molecule cu dipol permanent. Acestea sunt interactiuni puternice, iar separarile bazate pe ele se vor produce în conformitate cu polaritatea componentelor. Daca între moleculele fazei stationare si moleculele componentei exista forte de atractie puternice, se vor înregistra abateri negative de la legea lui Raoult, adica presiunea de vapori a componentei va fi mai mica decât cea prescrisa de aceasta lege (γ<1). Drept urmare, concentratia componentei în faza mobila va fi mai mica si componenta va fi puternic retinuta de faza stationara. Daca abaterea de la legea lui Raoult este pozitiva (γ>1), ceea ce se întîmpla în cazul compusilor cu polaritate diferita de cea a fazei stationare, presiunea de vapori a componentei va fi mai mare, concentratia sa in faza mobila de asemenea mai mare decât cea prescrisa de aceasta lege si în consecinta componenta va fi slab retinuta de faza stationara.
Interactiuni de inductie, care se manifesta între un dipol permanent si unul indus, care poate fi sau molecula fazei stationare sau molecula componentei analizate. Separarile bazate pe forte de inductie se vor produce în conformitate cu polarizabilitatea moleculei dipolului indus. Exemple de asemenea molecule sunt cele ale compusilor aromatici.
Interctiuni chimice, bazate pe formarea unor legaturi chimice reversibile între între moleculele fazei stationare si moleculele compusului analizat (de exemplu legaturile dintre hidrocarburile nesaturate si ionii Ag+).
Nu numai natura fazei stationare lichide are importanta asupra separarii ci si concentratia ei, adica încarcarea fazei suportului cu faza stationara. Aceasta încarcare influenteaza valoarea factorului de capacitate k', care creste odata cu cresterea cantitatii de faza stationara din coloana. Pe de alta parte însa, eficacitatea de separare a coloanei scade odata cu cresterea concentratiei de faza stationara, datorita cresterii înaltimii echivalente a talerului teoretic (creste valoarea parametrului df din termenul C din ecuatia Van Deemter, df fiind diametrul mediu al filmului de lichid, considerat uniform, care este depus pe suport). Se considera ca este mai indicat sa se foloseaca coloane având concentratii mai reduse de faza stationara pentru a evita scaderea performatelor coloanei. si aceasta reducere este însa limitata, pentru ca filmul de faza stationara sa poata acoperi toata suprafata suportului si a nu se înregistra adsorbtia componentelor analizate pe suport. În cazul coloanelor clasice, cu umplutura, concentratia de faza stationara cea mai indicata este în jur de 10%, pentru suporturile de tip Chromosorb W sau P. În cazul coloanelor capilare, grosimea filmului de faza stationara este cuprinsa în mod uzual între 0,1-1 μm.
Clasificarea fazelor stationare lichide se face în urmatoarele categorii principale:
Faze stationare nepolare, care sunt compusi de tip hidrocarburi (parafine) sau uleiuri siliconice (polisiloxani) care nu au grefate grupari polare. Exemple de asemenea faze stationare sunt: squalan (hidrocarbura C30H62), uleiuri siliconice de tip metilsilicon (denumiri comerciale OV-1, SE-30). Aceste faze separa compusii analizati în ordinea cresterii punctelor de fierbere ale acestora.
Faze stationare polare, care contin o proportie ridicata de grupari polare, de exemplu: polietilenglicoli cu masa moleculara medie (Carbowax 20M), uleiuri siliconice cu grupari cianopropil (OV-225 metil-fenil-cianopropil-silicon), dietilenglicolsuccinat (DEGS), esterul nitrotereftalic al polietilenglicolului (FFAP), etc. Ei diferentiaza compusii polari de cei nepolari, retinându-i numai pe cei polari. Se utilizeaza mai ales pentru separarea compusilor polari.
Faze stationare de polaritate intermediara, care contin grupari polare în concentratie mai mica sau grupari polarizabile, grefate pe un schelet nepolar. Exemple de astfel de faze sunt cele metil-fenil-siliconice (OV-17), dinonilftalatul, polietilenglicolii cu mase moleculari mari. Sunt faze stationare universale, care se pot folosi pentru analiza atât a compusilor polari cât si a celor nepolari.
Faze stationare specifice, care se utilizeaza în anumite cazuri particulare. Ele contin compusi care interactioneaza numai cu anumite componente ale amestecului de analizat, de exemplu AgNO3 dizolvat în polietilenglicol care formeaza aducti cu olefinele.
Faze stationare chirale, care contin compusi chirali ce interactioneaza doar cu un singur izomer optic al unei perechi de enantiomeri. Asemenea faze sunt cele pe baza de ciclodextrina sau anumiti aminoacizi.
Ţinând cont de polaritatea fazelor stationare si de interactiunile posibile, compusii organici pot fi grupati din punct de vedere al separarii cromatografice în urmatoarele cinci clase:
Clasa I - compusi foarte polari, capabili sa dea legaturi de hidrogen: apa, glicerina, glicoli, hidroxiacizi, aminoacizi. Acesti compusi sunt greu de separat prin cromatografie de gaze, din cauza polaritatii foarte mari. Cu exceptia apei, ei se derivatizeaza înainte de separare.
Clasa II - compusi polari, care au atomi de hidrogen activi: alcooli, acizi carboxilici, fenoli, amine primare si secundare, nitroderivati si nitrili cu un atom de hidrogen în pozitia α. Acesti compusi se separa pe faze stationare polare.
Clasa III - compusi de polaritate intermediara, care nu au hidrogen activ: eteri, esteri, aldehide, cetone, nitroderivati si nitrili fara atom de hidrogen în pozitia α. Acesti compusi se separa pe faze stationare de polaritate intermediara.
Clasa IV - compusi cu polaritate scazuta, care însa au hidrogen activ: hidrocarburi aromatice, alchene, cloroform, clorura de metilen, dicloretan, tricloretan, etc. Acesti compusi se separa pe faze stationare de polaritate intermediara sau nepolare.
Clasa V - compusi nepolari: alcani, cicloalcani. Acesti compusi se separa pe faze stationare nepolare.
Aceasta clasificare este empirica si are doar rolul de a usura alegerea fazei stationare celei mai potrivite pentru analiza cromatografica.
3.3.2.2. Alegerea fazei stationare. Constantele Mc Reynolds.
Asa cum s-a aratat, cel mai important criteriu pe baza careia se face alegerea fazei stationare pentru o anumita analiza este polaritatea fazei si a compusilor de analizat. Exprimarea numerica a acestei polaritati, care ar usura mult alegerea, nu este posibila pentru ca nu exista nici o marime fizica ce poate fi asociata direct cu polaritatea (în anumita masura momentul dipol ar putea fi o asemenea marime). Din acest motiv, pentru exprimarea polaritatii fazelor stationare se utilizeaza niste compusi de referinta, care se aleg în mod arbitrar. Un asemenea sistem a fost elaborat de Rohrschneider si dezvoltat apoi de Mc Reynolds, având la baza indicii de retentie Kovats si utilizând squalanul drept faza stationara de referinta careia i se atribuie polaritatea nula. Au fost selectati un numar de 5 compusi de referinta: benzen, 1-butanol, 2-pentanona, nitropropan si piridina. Fiecare dintre acesti compusi poate fi considerat ca etalon pentru anumite clase de substante, cu care se aseamana în ce priveste comportarea cromatografica, astfel:
benzenul, pentru hidrocarburi nesaturate si hidrocarburi aromatice;
1-butanolul, pentru alcooli, fenoli, acizi carboxilici;
2-pentanona, pentru aldehide, cetone, eteri, esteri;
nitropropanul, pentru nitroderivati si nitrili;
piridina, pentru baze aromatice si heterocicli cu azot.
La determinarea constantelor Mc Reynolds pentru o anumita faza stationara A, în cazul coloanelor cu umplutura, se procedeaza astfel:
fiecare compus de referinta se analizeaza pe o coloana cu 20% faza stationara squalan, izoterm la 100˚C si se determina indicii de retentie corespunzatori;
compusii de referinta se analizeaza pe o coloana care contine 20% faza stationara A, în aceleasi conditii si se determina si în aceste cazuri indicii de retentie Kovats;
se calculeaza constantele Mc Reynolds ale fazei stationare A, astfel:
pentru benzen (notat cu x'): diferenta dintre indicele de retentie al benzenului pe faza stationara A si pe squalan:
![]()
pentru 1-butanol (notat cu y'): diferenta dintre indicele de retentie al butanolului pe faza stationara A si pe squalan;
la fel se procedeaza pentru 2-pentanona (z'), nitropropan (u') si piridina (s').
Aceste valori ale constantelor Mc Reynolds au fost determinate pentru un numar mare de faze stationare uzuale si ele se gasesc tabelate. Câteva asemenea valori sunt date in tabelul 3.1.
Tabelul 3.1. Valoarea constantelor Mc Reynolds pentru unele faze stationare uzuale
|
Denumirea fazei stationare |
x' |
y' |
z' |
u' |
s' |
|
Squalan C30H62 |
0 |
0 |
0 |
0 |
0 |
|
Metilsilicon OV-1 |
16 |
55 |
44 |
64 |
42 |
|
Metilsilicon SE-30 |
15 |
53 |
44 |
64 |
41 |
|
Metil-fenil-silicon (20% fenil) OV-7 |
69 | ||||
|
Metil-fenil-silicon (50% fenil) OV-17 | |||||
|
Cianopropil-metil-fenil-silicon OV-225 | |||||
|
Polietilenglicol Carbowax 20M | |||||
|
Ester nitrotereftalic al PEG FFAP | |||||
|
Dietilenglicolsuccinat DEGS |
La ora actuala exista câteva sute de faze stationare utilizate în cromatografia de gaze si constantele Mc Reynolds permit selectarea aceleia care promite cea mai buna separare a componentelor analizate. Deoarece evident exista o serie de faze stationare care au valorile constantelor Mc Reynolds apropiate, acestea vor putea fi substituite între ele fara ca separarea sa fie afectata. Astfel, daca exista o faza stationara recomandata în literatura de specialitate pentru o separare dar care nu este disponibila într-un anumit laborator, ea va putea fi înlocuita cu una echivalenta.
Coloana reprezinta partea cea mai importanta a unui cromatograf, fiind sediul procesului de separare. Pentru ca separarea sa fie eficienta, trebuie sa fie alese în mod corespunzator fazele cromatografice (în cazul cromatografiei de gaze aceasta alegere se refera la faza stationara), dimensiunile coloanei, viteza fazei mobile si temperatura de analiza.
Coloanele cromatografice sunt de doua tipuri: clasice (cu umplutura) si capilare.
Au diametrul interior cuprins între 2 si 8 mm (uzual 4 mm) si lungimea între 0,5-3 m. Se pot confectiona din otel inoxidabil, sticla, mase plastice, iar forma coloanelor poate fi în forma de U (în cazul coloanelor scurte) sau spirala (Figura 3.2).

Figura 3.2. Coloana cromatografica cu umplutura
Coloanele confectionate din sticla au avantajul ca permit observarea vizuala a neregularitatilor de asezare a umpluturii (goluri, crapaturi) sau a eventualei deteriorari a umpluturii. Au însa dezavantajul ca din cauza friabilitatii sticlei conectarea la partile metalice ale aparatului este mai dificila. Coloanele din otel inoxidabil sunt cele mai utilizate. Ele se pot confectiona si în varianta preparativa, având lungimi cuprinse între 1-4 m si diametrul interior de 2,5 cm. Acestea permit analiza unor volume de proba mai mari de 1 ml.
Prepararea unei coloane clasice pentru cromatografia gaz-lichid presupune urmatoarele operatii:
depunerea fazei stationare;
umplerea coloanei;
conditionarea coloanei.
Depunerea fazei stationare se face prin cântarirea cantitatilor corespunzatoare de suport si faza stationara care urmeaza sa fie depusa (conform concentratiei pe suport stabilite), urmata de dizolvarea fazei stationare într-un solvent volatil potrivit (toluen, acetona, tetraclorura de carbon, etc.). Solutia de faza stationara se amesteca apoi cu suportul, iar din suspensia obtinuta se evapora încet solventul, într-un evaporator rotativ de vid, faza stationara ramânând atasata de suport.
Umplerea coloanei se face prin introducerea umpluturii obtinute prin depunerea fazei stationare pe suport la un capat al coloanei, la celalalt capat aplicându-se un vid nu foarte înaintat (trompa de apa, de exemplu). Evident ca la capatul la care se aplica vidul trebuie sa existe o sita sau alt opritor pentru ca umplutura sa ramâna în coloana. Pentru a favoriza asezarea compacta a umpluturii, coloana se bate sau se vibreaza în tipul umplerii. Controlul umplerii este mai usor de realizat în cazul coloanelor din sticla.
Conditionarea coloanei se realizeaza prin mentinerea coloanei timp de cel putin 10 ore la o temperatura situata cu aproximativ 5-10ºC mai jos decât temperatura maxima admisa pentru faza stationara respectiva. În timpul conditionarii capatul dinspre detector al coloanei nu se conecteaza la aparat, iar prin coloana trebuie sa treaca un debit mic de gaz purtator.
Drept coloane capilare sunt considerate acele coloane al caror diametru interior este mai mic de 1 mm (Figura 3.3). În mod obisnuit, ele se confectioneaza cu diametre de 0,25, 0,32 sau 0,53 mm, iar lungimea este în mod uzual cuprinsa între 15 si 50 m. Materialul de constructie cel mai utilizat este sticla, însa exista si coloane capilare confectionate din otel inoxidabil. În cazul coloanelor capilare, faza stationara se depune în strat foarte subtire pe peretele interior al coloanei, motiv pentru care aceste coloane sunt denumite si coloane tubulare deschise. Pentru cresterea uniformitatii si aderentei fazei stationare, peretele interior al tubului capilar se poate trata chimic, sau se poate depune un strat foarte subtire de suport.

Figura 3.3. Coloana capilara
Cea mai importanta operatie, de care depind in mare masura performantele coloanei, este depunerea peliculei de faza stationara pe peretele interior al tubului capilar. Aceasta depunere se poate face prin doua metode: metoda statica si metoda dinamica.
Metoda statica consta în umplerea coloanei cu o solutie ce contine cantitatea corespunzatoare (între 0,1-1%) de faza stationara dizolvata într-un solvent potrivit, în general clorura de metilen, dupa care se lasa sa se evapore solventul iar faza stationara ramâne atasata de peretele coloanei. Dezavantajul acestei metode este ca grosimea filmului de faza stationara nu este uniforma.
Metoda dinamica consta în trecerea unei solutii de faza stationara cu concentratia de aproximativ 10% în CH2Cl2 prin coloana, sub actiunea presiunii unui gaz inert pâna la umplerea completa a coloanei, dupa care lichidul se evacueaza tot sub actiunea presiunii de gaz iar o parte din faza stationara ramâne atasata de peretele coloanei. În continuare se sufla gaz inert prin coloana pâna la eliminarea completa a solventului si se determina cantitatea de faza stationara retinuta (prin cântarire).
Metoda utilizata pentru introducerea probei depinde de starea de agregare în care se gaseste proba: gazoasa, lichida sau solida. Este indicat în toate cazurile ca introducerea probei sa aiba loc într-un timp cât mai scurt. Marimea probelor variaza între mai putin de 1 μg în cazul coloanelor capilare si ordinul gramelor în cazul coloanelor preparative.
Probele gazoase sunt introduse cu ajutorul unor seringi speciale, rezistente la presiune, sau folosind dispozitive speciale care constau dintr-un robinet cu mai multe cai si o bucla calibrata (Figura 3.4). Aceste dispozitive permit introducerea unui volum cunoscut de proba în coloana printr-o singura miscare a rotorului robinetului, fara a se opri fluxul gazului purtator.
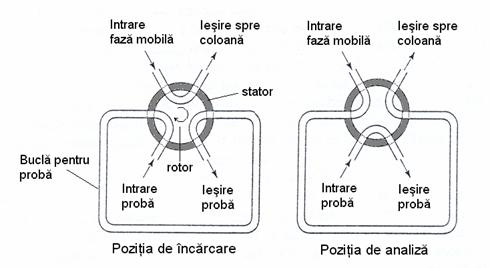
Figura 3.4. Dipozitiv pentru introducerea probei
În pozitia de încarcare, în coloana intra doar faza mobila, în timp ce proba gazoasa, care vine dintr-un recipient sub presiune, umple complet bucla calibrata iar excesul este evacuat în atmosfera. Dupa trecera rotorului pe pozitia de analiza, faza mobila intra în bucla unde preia proba, pe care o introduce în coloana. În acest timp fluxul gazos de proba este evacuat în atmosfera. Dupa terminarea analizei se trece din nou pe pozitia de încarcare si se introduce în bucla o noua cantitate de proba.
Probele lichide sunt introduse în coloana cu ajutorul unor microseringi, având în mod uzual capacitatea de 1, 5, sau 10 μl. Ele trebuie aduse în stare de vapori, ceea ce se realizeaza prin injectarea într-o camera de vaporizare (sau injector), care se gaseste înaintea coloanei si care este încalzita la o temperatura programata. Injectarea se face printr-un dop de cauciuc siliconic numit septum, care realizeaza izolarea injectorului de mediul exterior (Figura 3.5). Tot în camera de vaporizare se introduce si gazul purtator, care preia proba vaporizata si o transporta prin coloana.

Figura 3.5. Camera de vaporizare a unui cromatograf cu coloana capilara, prevazuta cu dispozitiv de divizare a probei (splitare)
Probele solide sunt introduse în coloana tot prin injectare cu microseringi, dupa ce au fost dizolvate într-un solvent adecvat. În cazul lor însa trebuie avut grija ca proba sa se vaporizeze la temperatura injectorului, altfel analiza nu va fi posibila, substantele cu punct de fierbere foarte rificat sau care se descompun înainte de fierbere neputând fi analizate prin cromatografie de gaze.
Exista cromatografe la care s-a renuntat la camera de vaporizare, injectarea probei realizându-se direct în partea superioara a coloanei, care este încadrata de o rezistenta de încalzire si joaca rolul injectorului. Aceasta tehnica se numeste on-column si cu ajutorul ei se elimina volumul mort corespunzator camerei de vaporizare, care poate influenta negativ analiza, mai ales atunci când volumul probei injectate este foarte mic.
În cazul coloanelor capilare se folosesc niste dispozitive de injectare speciale, care permit divizarea (splitarea) probei, cea mai mare parte fiind evacuata în atmosfera si doar o mica parte (de la 1/100 la 1/50 în mod uzual) fiind introdusa în coloana.
De asemenea, în cazul aparatelor moderne destinate analizei unui mare numar de probe se utilizeaza dispozitive de injectare automata (autosampler), care se pot atasa la cromatograf si permit analiza automata a unui mare numar de probe, fara interventia operatorului.
În cazul probelor care nu sunt stabile termic, au volatilitate scazuta sau prezinta picuri deformate cu coada (tailing), se procedeaza la transformarea lor chimica în derivati care sa nu mai aiba aceste inconveniente. Procedeul se numeste derivatizare si metodele aplicata cât si reactivii de derivatizare utilizati sunt de o mare diversitate, depinzând de natura compusului analizat. Una dintre metodele de derivatizare cele mai utilizate este silanizarea.
Detectorul reprezinta, dupa coloana cromatografica, cea mai importanta parte a unui cromatograf, el realizând punerea în evidenta a componentelor separate sub forma unor semnale electrice care pot fi integrate sau înregistrate.
Din punct de vedere al semnalelor pe care le emit, detectoarele pot fi de doua tipuri: integrale si diferentiale.
Detectoarele integrale emit semnale corespunzatoare masei totale a componentelor care ajung în detector, cromatogramele fiind niste curbe în trepte. Aceste tipuri nu mai sunt utilizate la ora actuala în practica cromatografica.
Detectoarele diferentiale emit semnale în functie de variatia unei anumite proprietati, pe masura ce componenta ajunge în detector, fiecarei componente corespunzându-i un semnal individual. Cromatograma se prezinta în acest caz ca o succesiune de picuri de elutie de tip gaussian, concentratia fiecarei componente fiind proportionala cu aria picului sau. Detectoarele diferentiale se pot clasifica la rândul lor în functie de sensibilitatea lor la concentratia sau la debitul de masa al compusului analizat.
În cazul detectoarelor cu raspuns la concentratia solutului în faza mobila aria picului solutului respectiv este data de relatia:
![]() (57)
(57)
unde m este masa totala a solutului care ajunge în detector, F este debitul fazei mobile, iar k1 este o constanta de proportionalitate. Se observa faptul ca aria fiind invers proportionala cu debitul fazei mobile, acest debit va trebui sa ramâna neaparat constant în timpul analizei pentru a nu avea erori, mai ales la analiza cantitativa. Din aceasta categorie fac parte detectoarele de conductivitate termica si cele cu captura de electroni.
În cazul detectoarelor cu raspuns la debitul de masa al solutului, aria picului va fi data de relatia:
![]() (58)
(58)
adica ea nu mai depinde de debitul fazei mobile si variatia acestuia în anumite limite nu mai reprezinta un factor perturbator al analizei. Din aceasta categorie fac parte detectoarele cu ionizare în flacara, cele cu spectrometru de masa si altele.
Dupa un alt criteriu, detectoarele se clasifica în universale si specifice. Detectoarele universale sunt sensibile la un numar mare ce compusi, în timp ce cele specifice sunt sensibile doar la anumiti compusi, respectiv la anumite grupe functionale sau legaturi din structura acestora. Dintre detectoarele universale se pot aminti detectorul de conductivitate termica, detectorul cu ionizare în flacara, detectorul cu spectrometru de masa, iar din categoria celor specifice detectorul cu captura de electroni, detectorul cu flacara alcalina (sau detectorul N(azot)-P(fosfor)), detectorul flamfotometric, etc.
3.4.1. Criterii de apreciere a performantelor detectoarelor
Principiul de constructie si functionare a detectoarelor fiind extrem de diferit, comparatia performantelor acestora este dificila. Exista totusi anumite criterii generale care permit o evaluare a performantelor unui detector.
a. Raspunsul detectorului
b. Sensibilitatea detectorului
Reprezinta variatia marimii semnalului emis de detector în functie de cantitatea de solut si se exprima prin valoarea pantei raspunsului detectorului pentru componenta respectiva (Figura 3.6). O sensibilitate mare a detectorului va duce la un raspuns mai mare pentru aceeasi cantitate de solut analizata si deci va permite analiza unor cantitati mici de proba, reprezintând o calitate importanta pentru un detector performant.

Figura 3.6. Sensibilitatea si liniaritatea raspunsului detectorului
c. Limita de sensibilitate
Este un criteriu strâns legat de cel precedent, definindu-se prin cantitatea minima din componenta respectiva pentru care se obtine un semnal de raspuns al detectorului de doua ori mai mare decât valoarea medie a zgomotului (semnalului) de fond (Figura 3.7). Cantitatea respectiva se numeste cantitate minima detectabila. Semnalul de fond reprezinta semnalul dat de detector în absenta vreunei componente injectate si depinde de caracteristicile sale constructive.

Figura 3.7. Limita de sensibilitate a detectorului
d. Domeniul de raspuns liniar
Reprezinta intervalul în care semnalul de raspuns al detectorului este direct proportional cu cantitatea de solut care ajunge în detector. Stabilirea acestei liniaritati se face prin etalonare, cu ajutorul unor etaloane pure ale compusilor analizati. Asemenea etaloane pentru cromatografia de gaze sunt accesibile comercial, la o puritate foarte avansata (în general peste 99,9%). Stabilirea domeniului de raspuns liniar este importanta mai ales pentru analiza cantitativa. În situatiile în care raspunsul nu este liniar, caz întîlnit mai ales la detectoarele folosite în cromatografia de lichide dar si la unele detectoare folosite în cromatografie de gaze, este necesara trasarea unor curbe de etalonare polinomiale pentru a se putea efectua analiza cantitativa.
e. Stabilitatea detectorului
Acest criteriu se refera la mentinerea liniei de baza fara deviatii, chiar în conditiile variatiei unor conditii de analiza: temperatura, presiunea si debitul fazei mobile. Cauzele care determina deriva liniei de baza (numita si drift) pot fi multiple si în general pot fi remediate.
Exista si alte conditii pe care trebuie sa le îndeplineasca un detector bun: viteza de raspuns cât mai mare, rezistenta ridicata la temperatura, simplitatea constructiei, pret cât mai scazut.
3.4.2. Detectorul de conductivitate termica (catarometru, TCD)
Este un detector universal, cu raspuns la concentratia solutului în faza mobila. Are avantajul ca este nedestructiv pentru proba, deci poate fi utilizat în cromatografia preparativa sau cuplat cu alte instrumente pentru analiza suplimentara a probei: spectrometru de masa, spectrofotometru în IR, etc.
Principiul sau de functionare se bazeaza pe dependenta pierderii caldurii de catre un fir metalic încalzit, în functie de compozitia mediului gazos înconjurator. El consta din doua filamente metalice, care se gasesc în câte un canal de forma cilindrica din interiorul unui bloc metalic ce poate fi încalzit la o temperatura programata (Figura 3.8). Unul dintre filamente este filamentul de masura, iar celalalt este filamentul de referinta.

Figura 3.8. Detectorul de conductibilitate termica
Prin fiecare filament trece un curent de aceeasi intensitate, determinând încalzirea sa la o anumita temperatura. Daca în spatiul înconjurator filamentului se gaseste numai gaz purtator, pierderea de caldura va fi constanta si aceeasi pentru ambele filamente. Daca însa în celula de masura ajunge o componenta eluata din coloana cromatografica, pierderea de caldura a filamentului de masura va fi diferita, deci si temperatura si rezistenta sa electrica se vor modifica. Cele doua filamente se gasesc într-un montaj electric de tip punte Wheatsone, iar modificarea rezistentei electrice a filamentului de masura va determina dezechilibrarea acestei punti si aparitia unui curent electric. Acest curent este amplificat, reprezentând semnalul se iesire al detectorului, iar intensitatea sa este este proportionala cu cantitate de componenta care a ajuns în detector.
Filamentele pot fi confectionate din wolfram, platina sau dintr-un aliaj wolfram-reniu si sunt pasivate pentru a reduce sensibilitatea lor la oxigen. Marimea semnalului detectorului depinde si de diferenta dintre conductibilitatea termica a gazului purtator si a componentei analizate. Valorile relative (raportate la heliu) ale acestei conductibilitati pentru câteva gaze permanente si compusi organici uzuali sunt prezentate în Tabelul 3.2:
Tabelul 3.2. Conductibilitatile termice relative ale unor gaze si substante organice
|
Denumirea compusului |
Conductivitatea termica relativa |
|
12,5 |
|
|
hidrogen | |
|
heliu | |
|
azot |
18 |
|
bioxid de carbon |
12,7 |
|
etan |
17,5 |
|
benzen |
9,9 |
|
cloroform |
6,0 |
|
acetat de etil |
9,9 |
Se observa ca numai hidrogenul si heliul vor putea fi folosite ca gaze purtatoare când se lucreaza cu acest tip de detecor, deoarece ele au cele mai mari valori ale conductibilitatii termice, iar semnalul detectorului pentru un anumit compus va fi cu atât mai mare cu cât diferenta dintre conductibilitatea termica a gazului purtator si a compusului analizat este mai mare.
Detectoarele de conductivitate termica au si unele dezavantaje;
- au sensibilitate mai mica decât alte tipuri de detectoare, cantitatea minima detectabila fiind în jur de 10-6-10-8 g.;
- domeniul de raspuns liniar este destul de redus;
- au volum mort relativ mare, ceea ce este un dezavantaj important mai ales când se lucreaza cu coloane capilare;
- timpul de raspuns este relativ mare comparativ cu alte detectoare.
La utilizarea practica a acestor detectoare trebuie tinut cont si de urmatoarele considerente:
- este foarte importanta mentinerea constanta a presiunii si debitului gazului purtator în timpul analizei;
- curentul filamentului trebuie întrerupt la schimbarea coloanei sau septumului, pentru a nu veni în contact cu oxigenul care ar determina distrugerea filamentului prin arderea sa;
- daca se constata o deviatie a liniei de baza ce nu poate fi eliminata, este posibil ca aceasta sa se datoreze depunerii pe filament a unor componente greu volatile. Îndepartarea acestora se poate face prin injectare de toluen sau xilen.
- nu se recomanda analiza cu acest tip de detector a halogenilor si derivatilor halogenati, deoarece acestia pot ataca filamentul.
3.4.3. Detectorul cu ionizare în flacara (FID)
Este tipul de detector cel mai mult folosit în cromatografia de gaze. Principul sau de functionare consta în masurarea modificarii conductibilitatii electrice a unei flacari de hidrogen în prezenta compusilor organici (Figura 3.9).

Figura 3.9. Detectorul cu ionizare în flacara
La iesirea din coloana cromatografica, gazul purtator împreuna cu componentele separate este amestecat cu un curent de hidrogen si este aprins la capatul unei mici duze de ardere. Aceasta duza se gaseste montata într-o camera de ardere în care se sufla un curent de aer pentru a asigura oxigenul necesar arderii. Camera de ardere se încalzeste la o temperatura programabila, folosind niste rezistente electrice care se gasesc în exteriorul camerei (nu sunt prezentate în figura). Detectorul are doi electrozi, dintre care unul este capacul duzei iar celalalt este electrodul colector care este pozitionat deasupra sau în jurul flacarii. Între cei doi electrozi se aplica o tensiune de polarizare, care este în general de 150 V. Drept urmare, prin flacara se stabileste un curent slab, de aproximativ 10-14A. La aparitia unui compus organic separat din coloana, acesta va ajunge în flacara si în urma arderii se vor genera specii ionice (mai ales ioni CHO+) si electroni liberi. Acestia vor fi colectati de electrozi si în consecinta se va produce cresterea intensitatii curentului, care poate ajunge pâna la 10-8A. Acest curent este amplificat într-un amplificator electronic si este înregistrat, reprezentând semnalul detecorului pentru componenta respectiva.
Duza de ardere poate fi constituita din diferite materiale: cuart, ceramica, otel inoxidabil, nichel. Este important ca diametrul sau sa fie mic, pentru ca flacara sa fie punctiforma.
Sensibilitatea detectorului depinde de numarul ionilor formati în urma arderii compusului respectiv. Ea este proportionala cu numarul atomilor de carbon din molecula compusului legati de alti atomi de carbon sau atomi de hidrogen, la care se adauaga contributia atomilor de carbon din legati de halogeni, grupari hidroxil, amino si altele. Atomii de carbon din gruparile carbonil si carboxil nu au nici o contributie la semnalul detectorului. Din acest motiv exista o serie de compusi care nu dau raspuns sau dau raspuns foarte slab în detectorul cu ionizare în flacara. Asemenea compusi sunt: H2O, H2S, SO2, CS2, N2O, NH3, CO, CO2, formaldehida, acid formic si altele. Aceasta lipsa de sensibilitate poate fi uneori avantajoasa în sensul ca pot fi analizate solutii apoase fara ca apa utilizata ca solvent sa interfere la detectie cu vreunul din compusii analizati.
Sensibilitatea detectorului FID depinde de raportul hidrogen/gaz purtator, având o valoare maxima la un raport ce depinde de constructia detectorului. Debitul de aer influenteaza de asemenea sensibilitatea, deoarece un debit prea mic duce la scaderea acesteia si mareste instabilitatea liniei de baza prin cresterea zgomotului de fond.
Detectoarele cu ionizare în flacara sunt în medie de 1000 de ori mai sensibile decât cele de conductivitate termica, cantitatea minima detectabila fiind cuprinsa între 10-9-10-12 g. Trebuie mentionat ca raspunsul detectorului pentru o anumita componenta depinde de structura acesteia, de aceea pentru a se realiza analiza cantitativa trebuie facuta neaparat etalonare.
Câteva aspecte practice de care trebuie tinut cont la folosirea detectoarelor FID sunt urmatoarele:
- nu se lucreaza la temperaturi ale detectorului mai mici de 100ºC;
- temperatura detectorului trebuie sa fie superioara temperaturilor de fierbere ale tuturor componentelor analizate;
- compusii aromatici lasa reziduuri pe detector, de aceea nu este recomandata folosirea lor ca solventi pentru probele supuse analizei;
3.4.4. Detectorul cu captura de electroni (ECD)
Face parte din categoria detectoarelor specifice, cu raspuns la concentratia probei. Principiul sau de functionare se bazeaza pe diferenta afinitatilor pentru electroni ale moleculelor diferitelor tipuri de substante. Afinitatea pentru electroni este determinata de proprietatea moleculelor substantelor organice în stare gazoasa de a aditiona electroni liberi, manifestându-se mai ales la compusii aromatici polinucleari, compusii care contin grupari carbonil conjugate, compusii organometalici, compusii organici halogenati, nitrili, etc. Ea nu se manifesta în schimb la alti compusi cum sunt hidrocarburile saturate. Asadar detectorul cu captura de electroni va fi foarte sensibil pentru anumite clase de compusi, putând detecta cantitati pâna de ordinul 10-9-10-13g si total insensibil pentru alti compusi.
Principiul de functionare al detectorului se bazeaza pe ionizarea moleculelelor gazului purtator, care este în general azotul, sub actiunea particulelor β produse de o sursa radioactiva, în general 63Ni.
![]()
Electronii liberi produsi în urma ionizarii au viteza foarte mare si ei nu se recombina cu ionii pozitivi, fiind colectati de un electrod aflat în camera de ionizare. Celalalt electrod al detectorului (catodul) este în general chiar celula care contine sursa radioactiva. Între cei doi electrozi se aplica un câmp electric slab, în general de 50V, iar în urma ionizarii moleculelor de azot prin iradiere se genereaza un curent electric slab, care este amplificat rezultând un semnal de baza al detectorului cu intensitatea de aproximativ 10-8A.
Daca în camera de ionizare a detectorului ajunge un compus organic cu afinitate mare pentru electroni, moleculele acestuia vor aditiona o parte din electroni formând ioni negativi, care însa având mobilitate mult mai mica decât electronii se vor putea recombina cu ionii pozitivi înainte de a fi colectati de electrod. Aceasta captura de electroni se poate produce în doua moduri: nedisociativ sau disociativ.
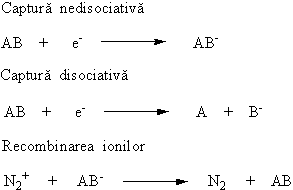
Datorita scaderii numarului ionilor care ajung la electrod, se produce reducerea intensitatii curentului electric. Asadar semnalul detectorului pentru compusul cu afinitate pentru electroni ar fi un pic negativ, dar pentru a opera în modul cunoscut de la celelalte detectoare acesta este transformat pe cale electronica într-un pic pozitiv.
Detectorul cu captura de electroni are avantajul selectivitatii si sensibilitatii ridicate, dar în acelasi timp si anumite dezavantaje:
- nu se pot utiliza decât anumite faze stationare, cu volatilitate foarte mica;
- electrodul se contamineaza repde, de exemplu cu urme de solventi clorurati;
- intervalul sau de liniaritate este foarte scurt;
- curatirea detectorului contaminat este mai dificila deoarece exista pericolul contaminarii radioactive a personalului. Ea se poate face în general prin încalzire la 350ºC sau spalare cu un solvent adecvat;
- gazul purtator si proba trebuie sa fie perfect uscate, deoarece si apa are proprietatea de a aditiona electroni.
Drept gaz purtator se mai poate folosi si argonul, cu continut de 5-10% metan. O atentie deosebita trebuie acordata mentinerii constante a temperaturii detectorului în timpul analizei, deoarece semnalul detectorului variaza foarte mult în functie de aceasta temperatura (de pîna la 5 ori la modificarea temperaturii cu 30ºC).
Cu toate dezavantajele sale, detectorul cu captura de electroni se foloseste foarte mult în anumite domenii, de exemplu pentru analiza compusilor halogenati toxici din apa sau a urmelor de substante antidaunatoare din produsele alimentare si din alte produse.
Daca o molecula este ionizata în vid, are loc fragmentarea sa cu formarea unui grup de ioni caracteristici cu diferite mase. Daca acesti ioni sînt separati printr-o metoda oarecare, reprezentarea abundentei lor în functie de masa constituie spectrul de masa. Spectrometria de masa a debutat la începutul secolului XX ca o metoda folosita de fizicieni pentru a pune în evidenta existenta izotopilor unor elemente, dar la ora actuala a devenit o metoda analitica de mare importanta într-o serie de domenii ale stiintei, incluzînd si chimia.
În principiu, spectrometria de masa consta din doua procese de baza:
- ionizarea moleculelor substantei de analizat urmata de fragmentarea si rearanjarea ionilor, care se realizeaza în camera de ionizare a spectrometrului.
- separarea ionilor formati în functie de masa lor si detectarea acestora, care are loc în analizorul de masa al spectrometrului.
3.5.1. Ionizarea moleculelor si fragmentarea ionilor
Ionizarea se poate efectua în doua modalitati: ionizare prin impact cu electroni si ionizare chimica.
3.5.1.1. Ionizarea prin impact cu electroni

Principiul
acestui proces poate fi urmarit pe baza schemei de constructie
si functionare a camerei de ionizare cu electroni, redat în Figura
3.10.
Figura 3.10. Camera de ionizare a spectrometrului de masa
În camera se realizeaza un vid înaintat, de 10-8 torr. Electronii proveniti de la un filament încalzit sunt focalizati în timp ce strabat camera si colectati de un electrod având un potential de 70 V. Acesta îi confera fiecarui electron o energie de 70 eV. Daca în camera de ionizare se introduce o substanta (de exemplu metanul) într-o cantitate suficienta ca presiunea sa creasca la 10-5 torr, ciocnirile dintre electroni si moleculele compusului respectiv determina fragmentarea acestora. Întrucât electronii la 70 eV au energie suficienta pentru a rupe orice legatura din molecula, prin fragmentare se vor forma toti ionii posibli, de exemplu în cazul metanului:
![]() CH4 + e CH4+,
CH3+, CH2+, CH+, C+
CH4 + e CH4+,
CH3+, CH2+, CH+, C+
Din cauza presiunii scazute din camera de ionizare, numai o molecula din aproximativ 106 va realiza ciocnirea si ionizarea. Presiunea de 10-5 torr în prezenta probei este importanta pentru realizarea unor spectre de masa reproductibile. Drumul liber mijlociu al ionilor sau moleculelor la aceasta presiune este mai mare decât dimensiunea camerei de ionizare, care este data de relatia aproximativa:
![]() (cm)
(cm)
Aceasta înseamna ca fragmentele de molecule neutre si de ioni formati nu se vor ciocni între ei ci numai cu peretii camerei, fiind posibila asadar îndepartarea lor din camera de ionizare si colectarea ionilor de mase diferite în detector. Spectrul de masa obtinut în asemenea conditii va fi reproductibil si caracteristic pentru compusul respectiv. În aceasta idee trebuie tinut cont de faptul ca abundenta relativa a ionilor formati în urma impactului cu electronii depinde în afara de energia electronilor de ionizare si de temperatura la care are loc ionizarea.
Ionizarea prin impact cu electroni duce la fragmentarea moleculei si la obtinerea unui amestec complex de ioni a caror masa si abundenta relativa se poate utiliza pentru identificarea calitativa a compusilor respectivi. Primul ion care se formeaza în urma impactului este asa-numitul ion molecular, care are mare importanta în identificarea substantei deoarece are aceeasi masa cu masa moleculara a compusului respectiv. Din pacate însa în aceste conditii stabilitatea ionului molecular este asa de redusa (el se va fragmenta mai departe) încât abundenta sa relativa va fi foarte mica si identificarea de cele mai multe ori imposibila (a se vedea exemplul hexaclorbenzenului). În consecinta, a devenit necesara utilizarea unei tehnici de ionizare mai blânde prin care sa se obtina informatii despre masa moleculara. Aceasta tehnica a fost ionizarea chimica, introdusa în anul 1966.
3.5.1.2. Ionizarea chimica
În spectrometria de masa prin ionizare chimica, în sursa de ioni se introduce un anumit gaz numit gaz reactiv, în conditiile în care se mentine o presiune relativ ridicata, în jur de 1 torr. Acest gaz este ionizat sub actiunea electronilor, obtinând-se ioni reactivi care realizeaza ionizarea moleculelor probei de analizat. Acestea nu sufera decât în masura foarte mica ionizare directa, datorita concentratiei mari a gazului reactiv în comparatie cu proba. Pe de alta parte, probabilitatea ciocnirilor cu ionii reactivi este mare din cauza presiunii ridicate din sursa de ioni. Reactiile dintre ionii reactivi si moleculele compusului de analizat au loc cu energie mica în comparatie cu impactul direct cu electronii, deci abundenta ionului molecular fata de alte fragmente rezultate va fi mare si mecanismul de fragmentare va fi mult mai simplu (Figura 3.11).
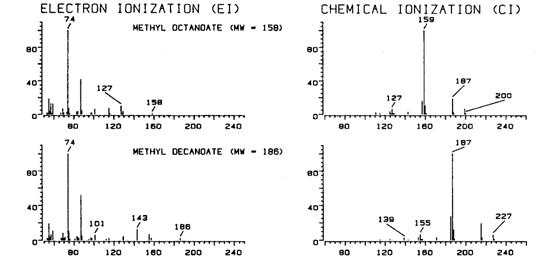
Figura 3.11. Spectrele de masa prin ionizare cu electroni si ionizare chimica cu metan
ale unor esteri
Majoritatea tehnicilor de ionizare chimica lucreaza cu ioni pozitivi, desi în ultima perioada s-au dezvoltat si cele cu ioni negativi (bazate pe captura unor electroni cu energie mai mica), iar gazul reactiv cel mai mult utilizat este metanul. Dezavantajul principal al tehnicilor de ionizare chimica este ca rezultatele depind foarte mult de conditiile de ionizare si nu este posibila alcatuirea unei librarii de spectre care sa fie folosita în scopuri de identificare, asa cum se procedeaza în cazul ionizarii prin impact cu electroni.
3.5.2. Separarea si detectarea ionilor
Separarea ionilor formati are loc într-un analizor de masa si este urmata de detectarea lor cu înregistrarea diagramei abundentei (concentratiei relative) în functie de masa ionilor, care reprezinta spectrul de masa.
În functie de metoda folosita pentru separarea ionilor, spectrometrele de masa pot fi de mai multe tipuri, cele mai uzuale fiind cele magnetice si cu cuadrupoli.
Utilizeaza un magnet pentru separarea ionilor proveniti din camera de ionizare, asa cum se poate observa în Figura 3.11.

Figura 3.11. Analizorul de masa
cu câmp magnetic si electric
Ionii care ies din sursa de ioni sunt accelerati cu ajutorul unui câmp electric cu o tensiune V cuprinsa între 2000 si 8000 V. În continuare fasciculul de ioni este focalizat si intra în sectorul magnetic al analizorului de masa unde este supus actiunii unui câmp magnetic de intensitate H, perpendicular pe directia sa de deplasare. Drept rezultat, ionilor li se imprima o miscare circulara cu raza de curbura r, devierea fasciculului fiind în cazul aratat în figura de 180º.
Ionii accelerati sunt separati sub influenta potentialului de accelerare si a câmpului magnetic, conform relatiei:
![]() (59)
(59)
unde m este masa ionului, z sarcina sa, H reprezinta intensitatea câmpului magnetic, r raza de curbura iar V tensiunea de accelerare aplicata ionilor. Deoarece majoritatea ionilor formati au sarcina z egala cu unitatea, la separare conteaza de fapt numai masa lor. Se observa ca pentru o anumita valoare a potentialului V si câmpului magnetic H numai ionii având o anumita valoare m/z se vor înscrie pe o traiectorie cu raza de curbura r si vor putea ajunge în detector. Din aceasta relatie mai rezulta ca masa ionului care trece prin analizor si ajunge în detector poate fi controlata prin modificarea potentialului V sau a câmpului magnetic H, deoarece raza de curbura r este o constanta data de constructia aparatului. În general se mentine potentialul de accelerare constant si se modifica câmpul magnetic pentru a realiza o scanare a întregului domeniu de masa. Dupa cum se observa din relatia (59), pe masura cresterii acestui câmp detectorul va înregistra ioni din ce în ce mai grei.
Detectorul de ioni al spectrometrului de masa este constituit în general dintr-un multiplicator electronic ce realizeaza atât detectarea cât si amplificarea curentului ionic datorat fragmentelor ionice care ajung în detector. Acest curent este apoi înregistrat, obtinându-se spectrul de masa.
Rezultatul analizei este asadar o diagrama ce contine reprezentarea concentratiilor relative (cunoscute sub numele de abundenta) ale fragmentelor ionice în functie de masa lor, mai exact raportul dintre masa si sarcina. Spectrele de masa pot fi înregistrate sub forma de picuri cu maxime, care constituie de fapt forma originala a spectrului, dar în practica se utilizeaza reprezentarea fiecarui pic sub forma unei linii (calculate pe baza spectrului original). Celei mai înalte asemenea linii din spectru i se acorda în mod arbitrar valoarea 100, iar restul sunt exprimate ca procentaje fata de aceasta, obtinându-se concentratiei diverselor fragmente ionice ca valori de abundenta relativa.
În figura 3.12 este prezentata schema de fragmentare a acetatului de etil, iar în Figura 3.13 spectrul de masa ala acestui compus în care se regasesc principalele benzi corespunzatoare fragmentelor ionice rezultate.
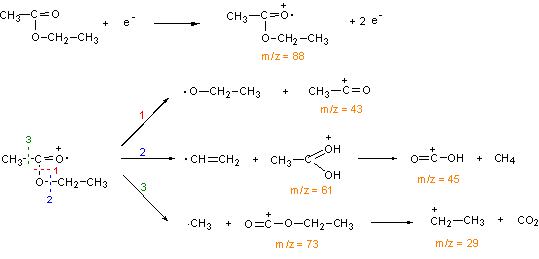
Figura 3.11. Schema de fragmentare a acetatului de etil

Figura 3.13. Spectrul de masa al acetatului de etil
3.5.3. Cuplarea cromatografului de gaze cu spectrometrul de masa
Atât cromatografia de gaze cât si spectrometria de masa sunt tehnici analitice de mare utilitate pentru analiza compusilor organici. Combinarea lor într-un singur sistem duce însa la obtinerea unor rezultate care depasesc mult ceea ce s-ar realiza prin simpla însumare a datelor oferite de cele doua tehnici. Pentru a putea gestiona imensa cantitate de date oferita de un sistem GC-MS este neaparat necesara utilizarea unui calculator echipat cu un program adecvat. Cuplarea cromatografiei de gaze cu spectrometria de masa ofera în primul rând posibilitatea identificarii calitative a componentelor analizate, cu o probabilitate de eroare foarte mica.
Optimizarea performantelor unui sistem GC-MS nu este însa deloc simpla, deoarece este vorba despre doua aparate care lucreaza în conditii mult diferite. Astfel, cromatograful de gaze foloseste diferite gaze purtatoare cu diverse debite, iar temperatura la iesirea din coloana si cantitatea din fiecare componenta variaza de asemenea pe un domeniu larg. Pe de alta parte, spectrometrul de masa lucreaza în conditii de vid avansat realizat în diferite variante, iar constructia camerei de ionizare si a analizorului de masa poate fi realizata în diverse modalitati. Problema principala este de a plasa o cantitate cât mai mare din fiecare compus organic, separat în coloana si corespunzator unui pic cromatografic, în camera de ionizare a spectrometrului fara a afecta vidul necesar functionarii acesteia. În aceasta camera presiunea trebuie mentinuta la o valoare sub 10-4 torr pentru a evita ciocnirile dintre ionii formati si molecule neutre, care ar duce la o fragmentare necontrolata. De asemenea, în analizorul de masa drumul liber mijlociu al moleculelor trebuie sa fie mai mare decât 200 cm pentru a nu avea loc ciocniri între ionii formati, iar acest lucru înseamna mentinerea unei presiuni mai scazute decât 10-5 torr.
Trecerea între cromatograf si spectrometru cu respectarea acestor conditii se realizeaza folosind niste dispozitive numite interfete.
3.5.3.1. Tipuri de interfete folosite în cromatografia GC-MS
Pe baza celor aratate, rezulta ca la trecerewa între cromatograf si spectrometru trebuie realizata o reducere a presiunii efluentului (constituit din gaz purtator si componenta separata) de aproximativ 108 ori, concomitent cu îndepartarea preferentiala a gazului purtator pentru ca în detector sa ajunga, pe cât posibil, numai moleculele componentei separate.
În cazul în care cromatograful este echipat cu coloana capilara (la care debitul fazei mobile este cuprins între 1-3 ml/min). În aceste conditii, daca spectrometrul de masa poseda pompe de vid de capacitate suficienta, toata cantitatea de efluent din coloana poate fi introdusa în sursa de ioni a spectrometrului. Aceste interfete se numesc directe, însa utilizarea lor prezinta inconvenientele ca schimbarea coloanei se poate face doar când spectrometrul de masa este oprit si cele doua instrumente nu se pot folosi independent una de alta. Pentru eliminarea acestor inconveniente au fost elaborate variante de interfete în care se face divizarea (sau splitarea) probei.
Dintre acestea, cea mai folosita este interfata deschisa cu splitare (Figura 3.14). Ea se caracterizeaza prin faptul ca portiunea terminala a coloanei capilare si o capilara mai subtire (cu diametrul de 0,15 mm) sunt îmbinate cu ajutorul unui manson de sticla, iar în spatiul înconjurator se sufla un curent de heliu, care preia excesul de proba ce nu intra în spectrometrul de masa. Divizarea probei se face în functie de diametrul capilarei care face legatura la spectrometrul de masa, cu cât aceasta este mai subtire raportul de splitare fiind mai mare. În acest mod, schimbarea coloanei cromatografice poate fi realizata fara a afecta functionarea spectrometrului de masa.
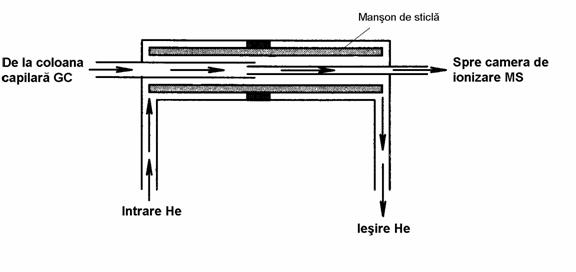
Figura 3.14. Interfata directa cu splitare
În cazul coloanelor cu umplutura nu se pot utiliza asemenea tipuri de interfete deoarece debitele de faza mobila sunt prea mari (uzual în intervalul 15-40 ml/min). Este necesara de aceea eliminarea celei mai mari parti din efluentul coloanei, concomitent cu concentrarea probei, dar fara a afecta compozitia ei. În acest scop se utilizeaza niste dispozitive numite separatoare moleculare, care realizeaza o reducere brusca a presiunii efluentului într-un spatiu situat între capatul coloanei cromatografice si camera de ionizare a spectrometrului de masa, concomitent cu eliminarea preferentiala a gazului purtator. Separarea se bazeaza pe marimile diferite ale moleculelor gazului purtator si componentei de analizat. Ea se poate realiza de exemplu prin trecerea preferentiala a moleculelor gazului purtator prin peretii unui tub de sticla poroasa în exteriorul careia se face vid, sau prin asezarea unei membrane din cauciuc siliconic la intrarea în camera de ionizare a spectrometrului, membrana care este permeabila doar pentru moleculele organice.
Sistemul cel mai utilizat de separator molecular este însa separatorul (interfata) cu jet (Figura 4.15). Moleculele gazului purtator si componentei separate în coloana trec dupa iesirea din coloana printr-o fanta îngusta într-un spatiu în care se realizeaza un anumit vid, având loc o crestere însemnata a vitezei de curgere. Moleculele componentei organice, mai mari si mai grele, vor avea tendinta de a se deplasa rectiliniu si vor fi directionate printr-o a doua fanta care se gaseste la aproximativ 0,5 mm de prima. În acelasi timp, moleculele gazului purtator care sunt mai usoare vor fi aspirate preferential de pompa de vid. Mai departe, prin acelasi mecanism are loc trecerea moleculelor din separator în camera de ionizare a spectrometrului de masa. Exista posibilitatea de a construi asemenea separatoare cu una, doua sau mai multe trepte. Separatorul din Figura 4.15 este construit în doua trepte. În treapta a doua procesul de separare are loc în mod identic, cu deosebirea ca presiunea trebuie sa fie mai scazuta decât în prima treapta.
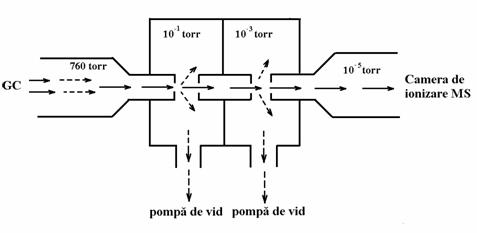
Figura 3.15. Separatorul molecular cu jet
3.5.3.2. Realizarea practica a analizelor GC-MS
Pentru utilizarea practica a sistemelor GC-MS pot fi luate în considerare doua alternative:
Fixarea spectrometrului de masa la o anumita valoare m/z caracteristica doar pentru o anumita componenta care ne intereseaza, el functionând în acest caz ca un detector foarte specific.
Parcurgerea (scanarea) întregului domeniu de masa într-un interval de timp redus, ceea ce înseamna ca se obtin sute sau chiar mii de spectre pentru fiecare analiza. Toate aceste spectre sunt stocate în memoria calculatorului atasat instrumentului.
În practica se utilizeaza cu precadere cea de-a doua varianta, pentru ca ea permite identificarea calitativa a tuturor componentelor prezente în amestecul supus analizei si nu doar a uneia dintre acestea. Acest lucru înseamna însa ca este necesara prelucrarea unui numar foarte mare de date, deoarece spectrometrele moderne au viteza de scanare ridicata (de obicei între 0,1-1,5 secunde). Apelarea oricarui dintre spectrele de masa înregistrate se poate face simplu, prin asociere cu numarul scanarii sau cu timpul trecut de la începutul analizei si care corespunde timpului de retentie obtinut în conditiile utilizarii ca detector a spectrometrului de masa. Trebuie însa precizat ca acest timp de retentie nu va fi identic cu cel obtinut cu un alt detector (de exemplu FID), din cauza caracteristicilor constructive diferite ale celor doua detectoare, chiar daca se utilizeaza aceeasi coloana si acelasi gaz purtator. Din acest motiv este necesar ca analiza GC-MS sa produca si o cromatograma pe care sa fie evidentiate picurile corespunzatoare componentelor separate, la fel ca în cazul celorlalte detectoare.
Aceasta cromatograma se realizeaza pe baza curentului ionic total, care reprezinta însumarea curentilor corespunzatori tuturor ionilor rezultati în urma fragmentarii compusului respectiv. Acest curent ionic total este calculat pentru fiecare spectru de masa corespunzator unei scanari, iar diagrama curentului ionic total în functie de timp (sau numarul de scanari) constituie cromatograma asociata analizei respective. Evident ca atunci când nu elueaza nici o componenta curentul ionic total va fi slab, corespunzator liniei de baza, iar pe masura ce numarul moleculelor din substanta respectiva care ajung în spectrometrul de masa creste el va creste de asemenea si va fi maxim în zona concentratiei corespunzatoare timpului de retentie al substantei respective (maximul picului cromatografic). Datorita faptului ca raspunsul detecorului pentru fiecare compus va fi diferit, rezulta ca si marimile reltive ale picurilor vor fi diferite comparativ cu aceeasi analiza realizata cu un alt tip de detector. Uneori este posibil ca în cromatograma realizata pe baza spectrelor de masa sa nu fie separate toate picurile care apartin unor componente cu rezolutie mica, pentru ca capacitatea de rezolutie a acestui tip de detector este mai mica decât a altora. Cu cât viteza de scanare este mai mare, rezolutia componentelor în cromatograma va fi si ea mai mare.
Viteza de scanare ridicata este importanta nu numai pentru a realiza o rezolutie buna a picurilor vecine ci si pentru a obtine spectre de masa cât mai fidele si reproductibile. Daca în timpul eluarii unei componente concentratia se schimba mai repede decât timpul necesar realizarii spectrului de masa, acest spectru va fi distorsionat si identificarea calitativa a compusului va fi îngreunata. Din acest motiv pentru identificarea calitativa este recomandat sa se utilizeze un spectru de masa selectat din zona de vârf a picului compusului respectiv.
În conditii de analiza date, spectrul de masa al unei molecule reprezinta o caracteristica specifica numai compusului respectiv si poate fi utilizat pentru identificarea sa calitativa. Aceasta identificare este însa foarte dificila (desi nu imposibila) de realizat prin interpretarea spectrului de masa pe baza fragmentelor de ioni pe care le contine si a abundentei relative a acaestora. O varianta mult mai simpla este compararea cu spectre de referinta ale unor compusi cu structura cunoscuta. Aceasta comparatie se poate face cel mai usor cu ajutorul calculatorului. Exista o serie de variante de cautare pentru identificarea spectrului de masa al unui compus necunoscut prin comparatie cu spectrele stocate în biblioteca de spectre a calculatorului. Eficienta acestei variante de identificare calitativa depinde de numarul de spectre de masa care se gaseste în biblioteca, complexitatea programului de cautare si viteza de lucru a calculatorului. Bibliotecile moderne de spectre contin zeci de mii de spectre de masa ale compusilor organici cei mai folositi si ele se livreaza în general odata cu programul care realizeaza achizitia si prelucrarea datelor provenite de la spectrometrul de masa. Rolul operatorului este însa de a evalua critic rezultatele analizei calitative oferite de calculator si de a le compara cu rezultatele unor analize paralele efectuate cu alte metode care pot oferi informatii despre compusul analizat.
|
